Талассемия – это генетическая болезнь крови, при которой, из-за мутации генов, образовывается недостаточное количество гемоглобина в организме и происходит деформация эритроцитов.
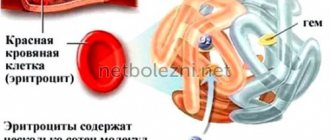
Гемоглобин – это специфический белок, который содержится в эритроцитах и отвечает за перенос железа и кислорода ко всем органам и тканям. Основная функция гемоглобина – дыхательная и его недостаток приводит к кислородному голоданию. В результате чего органы не могут полноценно выполнять свои физиологические функции.
Различают «взрослый» гемоглобин (HbA) и плодный тип гемоглобина (HbF).
Фетальный (плодный) гемоглобин синтезируется у плода спустя 2 недели после формирования внутренних органов (приблизительно с 12-й недели беременности). После рождения ребенка, HbF составляет до 80% и постепенно замещается «взрослым» HbA.
В норме у человека, плодного типа гемоглобина должно остаться не более 1,5%. Если случился сдвиг цифр влево или вправо – это всегда говорит о патологии (может быть нарушена работа сердечно-сосудистой системы, щитовидной железы, гипофиза, наличие гематологических отклонений, лейкоза, лимфогранулематоза, интоксикации).
Здоровая зрелая молекула гемоглобина (HbA) состоит из двух пар цепей – альфы и беты. Фетальный гемоглобин (HbF) состоит из цепей – гаммы и дельты.
А талассемия – это результат нарушенного синтеза одной или нескольких цепей гемоглобина. На основе чего различают альфа-талассемию, бета-талассемию, бета-дельту талассемию и т.д.
Тяжесть протекания того или другого вида заболевания зависит от количества поврежденных участков:
- Если изменен один ген цепи – заболевание (малая талассемия), протекает легко и бессимптомно. Но, при этом человек остается носителем данной болезни, которую может передать по наследству своим детям.
- Нарушение синтеза 2-х генов проявляется микроцитарной анемией в легкой степени тяжести.
- Патология 3-х генов протекает тяжело с ярко выраженной симптоматикой.
- Повреждение сразу 4-х генов (большая талассемия) – это наиболее редкий вид талассемии, который практически несовместим с жизнью (иногда ситуация решается пересадкой костного мозга).
Большая альфа-талассемия – самое серьезное отклонение, которое возникает еще в период внутриутробного развития плода. Грозит осложнениями, как для ребенка, так и для женщины.
Сегодня при своевременной внутриутробной трансфузии эритроцитарной массы удается сохранить жизнь плода.
Большая бета-талассемия (анемия Кули) – это также опасное нарушение, которое проявляется в первые два года жизни ребенка.
Малая альфа и бета-талассемия нарушает процесс эритропоэза (развитие новых эритроцитов), что приводит к хронической анемии и нарушению гемолиза.
Вне зависимости от вида, клиническое протекание талассемий схожи, различия зависят от степени тяжести патологического процесса.
Что такое талассемия и почему она возникает
Талассемия – это генетическое заболевание крови. Образуется недостаточное количество гемоглобина, белка, отвечающего за перенос кислорода и углекислого газа. Он состоит из четырех пептидов. 90% молекул гемоглобина содержат два альфа-глобина и два бета-глобина (гемоглобин А или A1), 2.5% содержат вместо бета-цепей дельта-цепи (гемоглобин А2), а остальные представляют собой гемоглобин A, состарившийся в процессе эксплуатации в эритроцитах (гемоглобин A3). При мутации в генах, отвечающих за синтез одного из глобинов, состав гемоглобина нарушается. Эти изменения влекут гибель эритроцитов.
Какие перспективы у «Зинтегло»?
Поскольку «Зинтегло» одобрен условно (без жёстких требований, как в случае других лекарственных средств), «Блюбёрд» обязана ежегодно отправлять регулятору обновленные клинические данные, чтобы тот продлевал регистрацию препарата, исходя из факта перевешивания пользы над вредом. Как только данные соберутся в пул должного объема, можно будет говорить о полноценном маркетинговом разрешении.
В дальнейшем к спектру пациентов, которым показана генотерапия «Зинтегло», будут добавлены больные с генотипом, характеризующимся полным отсутствием или критически малым эндогенным синтезом бета-глобина: например, β0/β0, мутации IVS-I-110 или IVS-I-5. Возможно, эффективность лечения таких пациентов менее выражена: проверка осуществляется в клинических испытаниях HGB-212.
В текущем году «Блюбёрд» намеревается отправить в Управление по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) регистрационное досье на «Зинтегло», запуск ожидается в 2020 году.
Попутно «Блюбёрд» завершает проверку «Зинтегло» в генно-терапевтическом лечении серповидно-клеточной анемии, поскольку эта патология вызвана иными мутациями того же гена бета-глобина.
Оценочно европейская популяция больных зависимой от гемотрансфузий бета-талассемией составляет 11–13 тыс. человек, причем половина проживает в Италии. Коммерческие перспективы в Соединенных Штатах гораздо скромнее с их пациентским пулом в пределах 1,4–1,5 тыс. человек. С учетом грядущего подключения серповидно-клеточной анемии для «Зинтегло» вполне реально заработать статус бестселлера, перешагнув порог в 1 млрд долларов годовых продаж.
Что касается конкуренции, в обозримом будущем свет увидит луспатерцепт (luspatercept): до начала декабря FDA примет решение относительно его одобрения в терапии бета-талассемии, зависимой от гемотрансфузий. Луспатерцепт, разрабатываемый «Акселерон фарма» (Acceleron Pharma) и «Селджен» (Celgene), представляет собой активатор созревания эритроцитов. Этот растворимый гибридный белок, включающий модифицированный внеклеточный домен активинового рецептора типа IIB (ActRIIB) и Fc-домен иммуноглобулина человека IgG1, таргетирован на определенные лиганды суперсемейства трансформирующего фактора роста бета (TGF-β), регулирующие эритропоэз на поздней стадии. Считается, что луспатерцепт работает как лигандная ловушка, сдерживающая патологическую сигнализацию Smad2/3, ответственную за неэффективный эритропоэз.
Причины и факторы риска развития заболевания
Талассемия – заболевание наследственное, то есть мутация передается от родителя ребенку. Если кто-то из родственников страдал от этой патологии, риск развития заболевания повышается.
Второй фактор риска – этническая принадлежность. Больше всего талассемия распространена в Африке, Средней Азии и странах Средиземноморья, где она и была открыта (в переводе с греческого “талассемия” — это “морская анемия”). Различают два основных типа талассемии: альфа и бета. В первом случае мутация затрагивает гены, отвечающие за синтез альфа-глобинов, во втором – бета.
Альфа-глобины кодируются четырьмя генами. Тяжесть заболевания будет зависеть от количества патологически измененных участков ДНК:
- 1 измененный ген – бессимптомная форма. Но при ней человек становится носителем заболевания и может передать его своим детям;
- 2 гена – легкое течение болезни;
- 3 гена – тяжелое течение болезни;
- 4 гена – редкий тип заболевания, который плохо совместим с жизнью. Большинство плодов гибнет еще в период внутриутробного развития, а родившиеся малыши, в основном, умирают вскоре после рождения или нуждаются в пожизненной терапии. В отдельных случаях удается их вылечить путем пересадки костного мозга.
Бета-глобины кодируются одним геном, который локализуется в 11-й хромосоме. Если дефектный ген содержится только в одной хромосоме из пары, заболевание протекает легко (малая талассемия). Повреждение обеих хромосом приводит к очень серьезному заболеванию, известному как большая талассемия или болезнь Кули (анемия Кули).
Частота встречаемости
Талассемия наследуется аутосомно-рецессивно. Это значит, что поражает одинаково и мальчиков, и девочек. Рецессивный характер говорит о том, что дети больные талассемией появляются в тех семьях, где мама и папа оба являются носителями мутаций. Хотя они могут даже не догадываться о своём носительстве, так как симптомы порой бывают незаметны.
В среднем частота встречаемости составляет 1 на 100 000 человек и может изменяться в зависимости от региона. Бета-талассемия встречается чаще, чем альфа.
Симптомы и признаки талассемии
В большинстве случаев талассемию определяют еще на этапе дородовой диагностики. При необходимости лечение начинают сразу, не дожидаясь появления симптомов. Если заболевание не выявила пренатальная диагностика, ожидаются следующие симптомы:
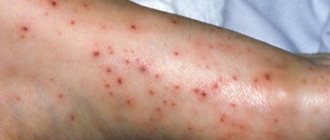
- бледность или желтушность слизистых оболочек;
- замедленный рост;
- темная моча;
- увеличение живота;
- деформация костей, особенно костей черепа.
Время появления первых признаков талассемии во многом зависит от типа заболевания и количества мутаций. У одних детей симптомы регистрируются вскоре после рождения, у других – в первые два года жизни.
Диагностика талассемии
Симптомы при талассемии бывают более или менее характерными. Чтобы поставить окончательный диагноз, врачу необходимы результаты лабораторных исследований. Обязателен при подозрении на талассемию общий анализ крови. Он покажет сниженное количество эритроцитов мелких, светлых, разных по форме и размеру. Кроме зрелых клеток, в мазке будет немало их предшественников — бластов. Дополнительно могут назначить другие специфические анализы крови для определения степени тяжести нарушений (биохимический анализ, определение железосвязывающей способности плазмы или ферритина в сыворотке). Также разработаны молекулярные тесты (ПЦР), позволяющие определить наличие мутаций.
Для оценки состояния печени, селезенки используют УЗИ, а для выявления патологии костной ткани – рентгенографию.
У ребенка талассемия может быть диагностирована еще на этапе вынашивания. Это исследование особенно рекомендуется проводить родителям, которые больны или могут быть носителями этого заболевания. Существует два метода диагностики:
- биопсия ворсинок хориона – проводится на 11-ой неделе беременности;
- амниоцентез (отбор околоплодных вод) – назначают на 16-й неделе.
Лечение талассемии
Определяется типом и степенью тяжести. При умеренно выраженных симптомах лечение не назначают. Время от времени проводят только переливание крови. В основном это нужно после операций, родов или для предотвращения возможных осложнений. Люди с бета-талассемией требуют более частых переливаний крови. Для нормализации избыточного уровня железа им также назначают специфические препараты, которые связывают и выводят железо.
При выраженной и тяжелой формах болезни существует два способа лечения:
- частые переливания крови (раз в несколько недель), которые сочетают с приемом препаратов, выводящих лишнее железо из организма;
- пересадка костного мозга – единственный метод, который помогает полностью излечить человека от талассемии. К сожалению, далеко не всегда трансплантация бывает успешной.
Лекарственные препараты при талассемии назначают только для коррекции симптомов и осложнений. Медикаментозной терапии самого заболевания не существует.
Сколько стоит «Зинтегло»?
P. S. «Блюбёрд» выставила стоимость «Зинтегло» в размере 1,575 млн евро (1,77 млн долларов). Как и предполагалось, оплата будет осуществляться в рассрочку на протяжении пяти лет равными долями: вначале аванс, а затем ежегодные платежи, но только в том случае, если зафиксирована результативность лечения.
«Блюбёрд» еще не определилась с конечной стоимостью своей генотерапии, но она, очевидно, окажется запредельной. Понятное дело, в высокоразвитых странах со страховой медициной никто из пациентов не платит всю сумму из собственного кармана.
Да, по подсчетам «Блюбёрд», «справедливая цена» «Зинтегло», рассчитанная с учетом улучшения качества и продолжительности жизни пациента, находится в пределах 2,1 млн долларов, но, как ожидается, итоговая стоимость будет существенно меньше. Указанная астрономическая сумма исходит, очевидно, из тех совокупных затрат системы здравоохранения на многолетнюю хроническую терапию бета-талассемии. Так, по оценкам экспертов, один американский пациент с этим заболеванием обходится где-то в 200–300 тыс. долларов ежегодно.
Предложено в первый год внести 20% авансом, а оставшуюся сумму выплачивать 20-процентным частями в рассрочку ежегодно в течение четырех лет, причем при условии, что эффективность лечения удовлетворяет предопределенным критериям, таким как исчезновение необходимости в переливаниях крови или существенное снижение числа таких процедур.
Другими словами, платить придется лишь по факту результативности генотерапии, тем более пока нет неоспоримых данных о продолжительности сохранения ее целительного действия. Подход кажется разумным, если принимать во внимание различия в промежуточных исходах среди больных, хотя общая эффективность «Зинтегло» и правда впечатляет.
Согласно прогнозам отраслевых экспертов, стоимость «Зинтегло» составит 1,2 млн долларов в США и 900 тыс. долларов (785 тыс. евро) в Европе.
Осложнения талассемии
Возможные осложнения заболевания:
- излишек железа, которое входит в состав гемоглобина. Накопление этого элемента в организме приводит к поражению сердца, печени, эндокринной системы;
- подверженность инфекциям. Особенно актуально для пациентов, которым удалили селезенку;
- деформация костей, связанная с увеличением объема костного мозга. Чаще всего этот процесс затрагивает кости черепа, реже конечностей. Они истончаются и чаще ломаются;
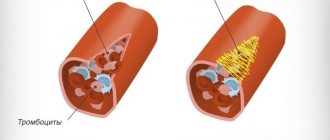
- спленомегалия — увеличение селезенки, где в основном и гибнут дефектные эритроциты. Если селезенка увеличена очень сильно, ее удаляют. Эта операция называется спленэктомия;
- задержка роста и полового созревания;
- сердечные болезни (хроническая сердечная недостаточность и аритмии) могут развиться при тяжелом течении заболевания.