Как проявляется талассемия?
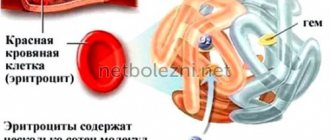
- В первую очередь, для талассемии характерна тяжелая форма анемии – снижение количеств гемоглобина и эритроцитов и, следовательно, нарушение переноса кислорода и дыхания. Из-за анемии возникает слабость, головокружение, одышка.
- Кроме того, при талассемии организм нередко перегружен железом: ведь гемоглобина образуется меньше и атомы железа остаются в свободной форме в крови. Железо накапливается в органах и тканях и мешает им нормально функционировать. Прежде всего от этого страдают печень, сердце и эндокринные железы.
- Еще одно опасное последствие талассемии – увеличение размеров селезенки. Именно в этом органе происходит распад эритроцитов, который повышается при болезни. Чем больше селезенка, тем больше риск ее разрыва, который опасен для жизни пациентов.
- Также при талассемии часто деформируются кости. Это происходит потому, что из-за сниженного количества эритроцитов в костном мозге расширяются участки кроветворения. Клетки костного мозга даже могут выходить за пределы костей, образуя утолщения – псевдоопухоли. Это ведет не только к деформации скелета, но и к другим симптомам – ведь кости могут задевать нервы и сосуды.
Как железо влияет на самочувствие и здоровье
Мутации гемоглобина и тяжесть болезни
Гемоглобин состоит из двух типов цепей – альфа и бета. В соответствии с геном цепи, в котором произошла мутация, талассемии тоже делят на альфа- и бета-тип. При этом альфа-цепь гемоглобина у человека кодируют сразу две пары генов, а бета-цепь – всего одна. В зависимости от количества мутировавших аллелей, талассемии разделяют по тяжести.
Так, если перестает правильно работать лишь одна копия гена альфа-глобина, болезнь проходит почти бессимптомно. Нарушение работы двух копий ведет к легкой форме альфа-талассемии, трех – к клинически выраженным нарушениям (эта форма называется гемоглобинопатия Н). Если же неправильно работают все четыре копии гена альфа-глобина, чаще всего гибель происходит еще на этапе плода. Похожим образом происходит и развитие бета-талассемии. Если одна из двух копий гена кодирует функциональный бета-глобин – талассемия проходит в легкой форме. При наличии в обеих копиях гена мутаций, влияющих на выработку бета-глобина, наблюдается тяжелая форма болезни – талассемия Кули. А вот если нарушения происходят сразу и в альфа-, и в бета-цепи – образование гемоглобина компенсируется и болезнь проходит мягче. Дело в том, что негативный эффект мутаций в генах цепей гемоглобина состоит не только в снижении количества белка, переносящего кислород, но и в снижении равновесия между двумя типами цепей. Из-за этого поломка лишь в гене альфа-глобина может оказаться даже опаснее, чем сразу в двух типах генов. Однако, конечно, такие сочетания встречаются крайне редко.
Железодефицитная анемия: кто в группе риска и как предотвратить болезнь?
Публикации в СМИ
Талассемия — гипохромная микроцитарная анемия с наследуемыми аномалиями генов глобинов. Талассемия распространена в средиземноморских странах, на Среднем Востоке и в Южной Азии.
Типы
• a-Талассемия вызвана дефектом синтеза a-цепи глобина. Преобладает в азиатских странах. Для окончательного диагноза необходимо провести детальный и углублённый анализ цепей Hb. У новорождённых при электрофорезе крови из пуповины находят Hb Барта. Содержание HbA2 и HbF обычно не увеличено •• Делеции четырёх генов. Неспособность продуцировать ни одной a-глобиновой цепи приводит к избытку g-глобиновых цепей, образующих тетрамеры Hb, называемого Hb Барта. Hb Барта имеет сильное сродство к кислороду, что препятствует тканевому дыханию — развиваются тяжёлая анемия, сердечная недостаточность, гепато- и спленомегалия, генерализованный отёк, и наступает внутриутробная смерть в результате водянки плода •• Делеции трёх генов (болезнь HbН, гемоглобинопатия Н). Несмотря на наличие выраженной анемии и повышенного содержания Hb Барта, образуется достаточное для развития плода количество a-глобина. В течение всей жизни у пациента сохраняется анемия, варьирующая от средней до тяжёлой. В постнатальном периоде преобладает HbH •• Делеции двух генов (малая талассемия). Умеренная гипохромная микроцитарная анемия. Иногда малую талассемию ошибочно диагностируют как ЖДА •• Делеция одного гена (состояние здорового носительства). Характерна нормальная картина периферической крови, включая нормальные концентрации Hb, Ht и количество эритроцитов. Патологию выявляют при количественном измерении глобиновых цепей или при анализе генома. Носитель может перенести обострение болезни HbH или малой талассемии.
• b-Талассемия (более 90% всех талассемий) развивается в результате экспрессии аномальных генов b-глобиновой цепи. Так как в геноме два аллеля b-глобина, существует две различные формы b-талассемии •• Гомозиготная b-талассемия (большая талассемия, анемия Кули) — тяжёлое заболевание, проявляющееся в детском возрасте и заканчивающееся летально к 20 годам; пациенты обычно трансфузиозависимы. Клинически проявляется задержкой роста, гепатоспленомегалией, незначительной желтухой, гиперплазией костного мозга и деформацией костей. Содержание HbA2 снижено или повышено, HbF — значительно повышено •• Гетерозиготная b-талассемия (малая талассемия) — обычно умеренная анемия, больные не зависят от трансфузий. Малая талассемия распространена в Италии и Греции. В равнинных областях Азербайджана ею страдает 7–11% населения. Содержание HbA2 повышено, HbF в норме или слегка повышено.
Генетические аспекты • a-Талассемии (*141800, 16p, дефекты генов HBAC, HBA1, HBA2, HbHR, Â) • b-Талассемии (*141900, 11p15.5, Â).
Патогенез • Избыток непарных глобиновых цепей индуцирует образование нерастворимых тетрамеров, абсорбирующихся на мембранах эритроцитов и повреждающих их. Эритроидные клетки становятся чувствительными к разрушению фагоцитами костного мозга (отсюда дефектный эритропоэз) либо печени и селезёнки (отсюда гемолитическая анемия) • Отмечают относительную недостаточность фолиевой кислоты.
Клиническая картина • Гипохромная анемия • Вторичный гемохроматоз вследствие необоснованного применения препаратов железа и частых гемотрансфузий • Гемолитическая желтуха, холелитиаз и спленомегалия • Поражение суставов •• При большой талассемии — артрит голеностопных суставов, талалгия; возможно развитие вторичного подагрического артрита; костные деструктивные изменения в связи с нарушением кроветворения — «башенный» череп, седловидный нос •• При малой талассемии — короткие приступы синовиита крупных суставов без лихорадки, внесуставных симптомов и без развития деформаций • Дисплазии зубов.
Диагностика • Основные диагностические критерии — микроцитоз, гипохромия, пойкилоцитоз (эллиптоцитоз) • В зависимости от преобладания гемолиза или дефектного эритропоэза наблюдают снижение или повышение числа ретикулоцитов • Снижения количества эритроцитов обычно не происходит (важный дифференциально-диагностический признак) • Малая b-талассемия — увеличение концентрации HbА2 (5% по сравнению 2,5% в норме) • Большая b-талассемия — значительное увеличение фракции HbF.
Дифференциальная диагностика • ЖДА • Гемолитические анемии.
ЛЕЧЕНИЕ • Повторные переливания отмытых или размороженных эритроцитов • Дефероксамин 0,5–1 г/сут — при регулярном применении, образуя комплексное соединение с избытком железа, замедляет развитие гемосидероза • Фолиевая кислота 0,1–0,2 мг/сут • Антибиотикотерапия — при присоединении инфекции • Трансплантация костного мозга и спленэктомия — по показаниям.
Осложнения • Инфекции • Трофические язвы конечностей • Патологические переломы • Гемосидероз печени и сердца • Апластический или мегалобластный криз.
Синоним. Мишеневидноклеточная гемолитическая анемия
МКБ-10 • D56 Талассемия.
Наследование и эпидемиология
При этом в мире всего пять процентов населения – носители мутаций в генах цепей гемоглобина, а из них симптомам болезни подвержены чуть менее двух процентов людей. Стоит также учитывать, что жители разных регионов сталкиваются с талассемией с разной частотой. Прежде всего болезнь поражает жителей Средиземноморья, а также стран Центральной и Южной Азии. В России носителей мутаций, связанных с талассемией, сравнительно мало – около одного процента (для бета-типа).
Носители мутации могут даже не подозревать о своем статусе – часто повреждение лишь одной копии гена не ведёт к тяжелым симптомам. Однако носители мутации рискуют передать болезнь детям: если два носителя заведут ребенка, он может получить от родителей сразу обе копии мутировавшего гена с вероятностью около 25 процентов.
В предыдущем разделе мы уже обсуждали, что мутации в двух копиях генов приводит к более тяжелой форме талассемии. Но если посчитать вероятность такого события получится лишь около сотой процента.
Причины
Главная причина талассемии – наследственный фактор. При котором у одного пациента функционирует патологическая РНК (участвует в кодировании, чтении и регуляции генов), а у другого – ДНК (появляются изменения в хромосоме). На фоне этих изменений уменьшается или полностью прекращается синтез одной из цепочек Hb.
В норме количество альфа, бета-цепей – одинаково, поэтому изменение синтеза хотя бы одной из них приводит к дисбалансу и патологии.
Болезнь встречается как у девочек, так и у мальчиков.
Частота появлениябольшой талассемии 1:100 000, если оба родители носители мутационного гена.
Также на патогенез талассемии влияет и этническая принадлежность, так как чаще всего данная генетическая патология встречается у жителей Африки, Восточного Средиземноморья и Юго-Восточной, Центральной Азии. Определено это тем, что мутационный ген имеет большую сопротивляемость к малярии (обычно в африканских регионах) и большим количеством родственных браков, что приводит к разным генетическим нарушениям, включая талассемию.
В
Украине талассемия– редкая патология.
Всемирная организация здравоохранения приняла международную классификацию заболеваний 10-го пересмотра (мкб-10), как единый нормативный документ для ведения учета болезней, причин обращений пациентов в медицинские учреждения и причин смертности.
Согласно мкб талассемии (D56) имеет следующую классификацию:
- D56.0 – альфа-талассемия;
- D56.1 – бета-талассемия;
- D56.2 – дельта-бета талассемия;
- D56.3 – носитель талассемии;
- D56.9 – талассемия неуточненная.
Обновление кодов (мкб-11) будет вводиться в действие с 2022 года.
Диагностика талассемии
Самый точный способ диагностировать талассемию любой тяжести – исследование структуры ДНК. Установление последовательности ДНК позволит обнаружить нарушенные варианты генов и не только вовремя принять меры в случае болезни, но и планировать беременность в случае бессимптомного носительства.
Талассемию также можно диагностировать при помощи анализов крови: общего, биохимического, а также определения в крови фракции гемоглобина. Эти исследования позволяют определить количество эритроцитов, их объем, а также концентрацию гемоглобина и уровни некоторых других веществ, чтобы по полученным результататам определить тип талассемии. При подозрении на талассемию также проводят УЗИ органов брюшной полости, чтобы оценить размеры селезенки и печени, которые страдают при болезни.
Генетический тест Атлас помогает определить патогенные варианты в гене, отвечающем за альфа-талассемию, обнаружить риски, связанные с проявлением симптомов и передачей заболевания детям.
Лечени
Малая талассемия (нарушение одного гена) – не требует лечения. Достаточно здорового образа жизни, сбалансированного питания, физической активности и отсутствия пагубных привычек. Все это «поможет» организму самостоятельно компенсировать дефект гена.
Большая талассемия и средней степени тяжести нуждается в специальном лечении, которое в «Клинике Спиженко» включает:
- Трансфузионную терапию (переливание эритроцитарной массы);
- Хелатную терапию (выведение лишнего железа из организма);
- Спленэктомию (удаление селезенки);
Лекарственные препараты при талассемии назначаются только для коррекции симптомов и предупреждения осложнений. Медикаментозного лечения талассемии – нет.
Пациенты с талассемией средней тяжести в донорских трансфузиях нуждаются не часто, только в критических ситуациях для организма:
- При беременности;
- Сердечной недостаточности;
- Резком снижении уровня гемоглобина (меньше 50 г/л);
- Оперативном вмешательстве;
- Заболеваниях опорно-двигательного аппарата;
- Легочной гипертензии.
Терапия и профилактика
Для лечения тяжелых форм талассемии используют переливание крови – для повышения количества эритроцитов и восстановления кислородного транспорта, а также хелатирование – для выведения свободной формы железа, которая не встроилась в «неправильный» гемоглобин. При тяжелой форме бета-талассемии переливание крови необходимо часто – каждые 3-4 недели. Однако сейчас такая терапия позволяет пациентам вести нормальный образ жизни и восстанавливает продолжительность жизни. Для легких форм талассемии показания проще – врачи советуют соблюдать здоровый образ жизни и больше двигаться. Занятия спортом позволяют поддерживать минерализацию костей скелета и препятствуют их деформации.
На заметку
- Талассемия – тяжелое наследственное заболевание, возникающее из-за мутаций в генах гемоглобина. Для нее свойственны слабость и головокружения, болезни печени и сердца, увеличение размеров селезенки, а также деформация костей.
- Выделяют два вида болезни – альфа- и бета-талассемию. В среднем в мире носителей мутаций, характерных для обоих видов, – около пяти процентов, в России же – всего около одного процента.
- Диагностируют талассемию при помощи анализов крови, УЗИ и генетического теста.
- Для лечения тяжелых форм используют частые переливания крови и выведение свободных форм железа. Для легких форм оказывается достаточно поддержания здорового образа жизни.
Источники:
- Практическая медицина, Гемоглобинопатии и талассемические сидромы, 2015
- Subcellular Biochemistry, Hemoglobin: Structure, Function and Allostery, 2020
- ФГУ ФНКЦ ДГОИ Росздрава, Эпидемиология гемоглобинопатий в Москве, 2008
- American Family Physician, Alpha and Beta Thalassemia, 2009
- NCBI, Thalassemia, 2021