How does thalassemia manifest?
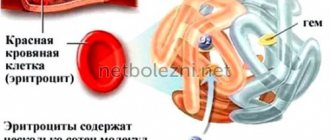
- First of all, thalassemia is characterized by a severe form of anemia - a decrease in the amount of hemoglobin and red blood cells and, consequently, a violation of oxygen transfer and respiration. Anemia causes weakness, dizziness, and shortness of breath.
- In addition, with thalassemia, the body is often overloaded with iron : after all, less hemoglobin is formed and iron atoms remain in free form in the blood. Iron accumulates in organs and tissues and prevents them from functioning normally. First of all, the liver, heart and endocrine glands suffer from this.
- Another dangerous consequence of thalassemia is an increase in the size of the spleen . It is in this organ that the breakdown of red blood cells occurs, which increases during illness. The larger the spleen, the greater the risk of rupture, which is life-threatening for patients.
- Also, with thalassemia, bones are often deformed . This occurs because, due to the reduced number of red blood cells in the bone marrow, the areas of hematopoiesis expand. Bone marrow cells can even extend beyond the bones, forming thickenings called pseudotumors. This leads not only to skeletal deformation, but also to other symptoms - after all, bones can touch nerves and blood vessels.
How iron affects well-being and health
Hemoglobin mutations and disease severity
Hemoglobin consists of two types of chains - alpha and beta. According to the gene chain in which the mutation occurred, thalassemias are also divided into alpha and beta types. At the same time, the alpha chain of hemoglobin in humans is encoded by two pairs of genes, and the beta chain is encoded by only one. Depending on the number of mutated alleles, thalassemias are divided according to severity.
So, if only one copy of the alpha-globin gene stops working correctly, the disease is almost asymptomatic. Disruption of two copies leads to a mild form of alpha thalassemia, three - to clinically significant disorders (this form is called hemoglobinopathy H). If all four copies of the alpha globin gene do not work correctly, most often death occurs at the fetal stage. The development of beta thalassemia occurs in a similar way. If one of the two copies of the gene encodes functional beta globin, thalassemia is mild. If there are mutations in both copies of the gene that affect the production of beta globin, a severe form of the disease is observed - Cooley's thalassemia. But if disturbances occur simultaneously in both the alpha and beta chains, the formation of hemoglobin is compensated and the disease passes more mildly. The fact is that the negative effect of mutations in the genes of hemoglobin chains is not only a decrease in the amount of oxygen-transporting protein, but also a decrease in the balance between the two types of chains. Because of this, a breakdown in only the alpha-globin gene can be even more dangerous than in two types of genes at once. However, of course, such combinations are extremely rare.
Iron deficiency anemia: who is at risk and how to prevent the disease?
Publications in the media
Thalassemia is a hypochromic microcytic anemia with inherited abnormalities of globin genes. Thalassemia is common in Mediterranean countries, the Middle East and South Asia.
Types
• a-Thalassemia is caused by a defect in the synthesis of the a-chain of globin. Predominant in Asian countries. For a final diagnosis, it is necessary to conduct a detailed and in-depth analysis of Hb chains. In newborns, during electrophoresis of blood from the umbilical cord, Barth's Hb is found. The content of HbA2 and HbF is usually not increased • • Deletions of four genes . Failure to produce any a-globin chains results in an excess of g-globin chains forming Hb tetramers, called Bart's Hb. Barth's Hb has a strong affinity for oxygen, which interferes with tissue respiration - severe anemia, heart failure, hepato- and splenomegaly, generalized edema develop, and intrauterine death occurs as a result of fetal hydrops • • Deletions of three genes (HbH disease, hemoglobinopathy H). Despite the presence of severe anemia and increased levels of Hb Bart, a sufficient amount of a-globin is formed for fetal development. The patient continues to have anemia throughout his life, varying from moderate to severe. In the postnatal period, HbH predominates • • Deletions of two genes (thalassemia minor). Moderate hypochromic microcytic anemia. Thalassemia minor is sometimes misdiagnosed as IDA • • Single gene deletion (healthy carrier state). A normal picture of peripheral blood is characteristic, including normal concentrations of Hb, Ht and the number of red blood cells. Pathology is detected by quantitative measurement of globin chains or by genome analysis. The carrier may experience an exacerbation of HbH disease or thalassemia minor.
• b-Thalassemia (more than 90% of all thalassemias) develops as a result of the expression of abnormal genes of the b-globin chain. Since there are two alleles of b-globin in the genome, there are two different forms of b-thalassemia • • Homozygous b-thalassemia (thalassemia major, Cooley's anemia) is a severe disease that manifests itself in childhood and ends fatally by the age of 20; patients are usually transfusion dependent. Clinically manifested by growth retardation, hepatosplenomegaly, mild jaundice, bone marrow hyperplasia and bone deformation. The content of HbA2 is reduced or increased, HbF is significantly increased • • Heterozygous b-thalassemia (thalassemia minor) - usually moderate anemia, patients are not dependent on transfusions. Thalassemia minor is common in Italy and Greece. In the lowland regions of Azerbaijan, 7–11% of the population suffers from it. The HbA2 content is increased, HbF is normal or slightly increased.
Genetic aspects of • a-Thalassemia (*141800, 16p, defects of the HBAC, HBA1, HBA2, HbHR, Â genes) • b-Thalassemia (*141900, 11p15.5, Â).
Pathogenesis • An excess of unpaired globin chains induces the formation of insoluble tetramers that are absorbed on the membranes of erythrocytes and damage them. Erythroid cells become sensitive to destruction by phagocytes of the bone marrow (hence defective erythropoiesis) or the liver and spleen (hence hemolytic anemia) • Relative deficiency of folic acid is noted.
Clinical picture • Hypochromic anemia • Secondary hemochromatosis due to unreasonable use of iron supplements and frequent blood transfusions • Hemolytic jaundice, cholelithiasis and splenomegaly • Joint damage •• With thalassemia major - arthritis of the ankle joints, thalalgia; possible development of secondary gouty arthritis; bone destructive changes due to impaired hematopoiesis - “tower” skull, saddle nose •• With thalassemia minor - short attacks of synovitis of large joints without fever, extra-articular symptoms and without the development of deformities • Dental dysplasia.
Diagnostics • The main diagnostic criteria are microcytosis, hypochromia, poikilocytosis (elliptocytosis) • Depending on the predominance of hemolysis or defective erythropoiesis, a decrease or increase in the number of reticulocytes is observed • A decrease in the number of erythrocytes usually does not occur (an important differential diagnostic feature) • b-thalassemia minor - an increase HbA2 concentration (5% compared to 2.5% normal) • b-thalassemia major - a significant increase in the HbF fraction.
Differential diagnosis • IDA • Hemolytic anemia.
TREATMENT • Repeated transfusions of washed or thawed red blood cells • Deferoxamine 0.5-1 g/day - with regular use, forming a complex compound with excess iron, slows down the development of hemosiderosis • Folic acid 0.1-0.2 mg/day • Antibiotic therapy - with infection • Bone marrow transplantation and splenectomy - according to indications.
Complications • Infections • Trophic ulcers of the extremities • Pathological fractures • Hemosiderosis of the liver and heart • Aplastic or megaloblastic crisis.
Synonym . Target cell hemolytic anemia
ICD-10 • D56 Thalassemia.
Inheritance and epidemiology
At the same time, only five percent of the world's population are carriers of mutations in the genes of hemoglobin chains, and of these, slightly less than two percent of people are susceptible to symptoms of the disease. It is also worth considering that residents of different regions experience thalassemia with different frequencies. The disease primarily affects residents of the Mediterranean region, as well as the countries of Central and South Asia. In Russia, there are relatively few carriers of mutations associated with thalassemia - about one percent (for the beta type).
Carriers of the mutation may not even be aware of their status - often damage to just one copy of the gene does not lead to severe symptoms. However, mutation carriers run the risk of passing the disease on to their children: if two carriers have a child, he can receive both copies of the mutated gene from his parents at once with a probability of about 25 percent.
In the previous section, we already discussed that mutations in two copies of genes lead to a more severe form of thalassemia. But if you calculate the probability of such an event, it turns out to be only about a hundredth of a percent.
Causes
The main cause of thalassemia is a hereditary factor. In which one patient has abnormal RNA (participates in coding, reading and regulation of genes), and another has abnormal DNA (changes appear in the chromosome). Against the background of these changes, the synthesis of one of the Hb chains decreases or completely stops.
Normally, the number of alpha and beta chains is the same, so a change in the synthesis of at least one of them leads to an imbalance and pathology.
The disease occurs in both girls and boys.
The incidence of thalassemia major is 1:100,000 if both parents are carriers of the mutation gene.
Ethnicity also influences the pathogenesis of thalassemia, since most often this genetic pathology occurs in residents of Africa, the Eastern Mediterranean and Southeast and Central Asia. This is determined by the fact that the mutation gene has greater resistance to malaria (usually in African regions) and a large number of consanguineous marriages, which leads to various genetic disorders, including thalassemia.
In
Ukraine, thalassemia is a rare pathology.
The World Health Organization has adopted the International Classification of Diseases, 10th revision (ICD-10), as a single normative document for keeping records of diseases, reasons for patient visits to medical institutions and causes of mortality.
According to the ICD thalassemia (D56) has the following classification:
- D56.0 – alpha thalassemia;
- D56.1 – beta thalassemia;
- D56.2 – delta-beta thalassemia;
- D56.3 – carrier of thalassemia;
- D56.9 – thalassemia, unspecified.
The code update (ICD-11) will be introduced in 2022.
Diagnosis of thalassemia
The most accurate way to diagnose thalassemia of any severity is to study the DNA structure. Establishing the DNA sequence will make it possible to detect disrupted gene variants and not only take timely measures in case of illness, but also plan pregnancy in the case of asymptomatic carriage.
Thalassemia can also be diagnosed using blood tests: general, biochemical, as well as determination of the hemoglobin fraction in the blood. These tests can determine the number of red blood cells, their volume, as well as the concentration of hemoglobin and the levels of some other substances in order to determine the type of thalassemia from the results obtained. If thalassemia is suspected, an abdominal ultrasound is also performed to assess the size of the spleen and liver, which are affected by the disease.
The Atlas genetic test helps identify pathogenic variants in the gene responsible for alpha thalassemia, detect risks associated with the manifestation of symptoms and transmission of the disease to children.
Treatment
Thalassemia minor (single gene disorder) does not require treatment. A healthy lifestyle, a balanced diet, physical activity and the absence of bad habits are enough. All this will “help” the body independently compensate for the gene defect.
Thalassemia major and moderate severity require special treatment, which at the Spizhenko Clinic includes:
- Transfusion therapy (transfusion of red blood cells);
- Chelation therapy (removal of excess iron from the body);
- Splenectomy (removal of the spleen);
Medicines for thalassemia are prescribed only to correct symptoms and prevent complications. There is no drug treatment for thalassemia.
Patients with moderate thalassemia do not often need donor transfusions, only in critical situations for the body:
- During pregnancy;
- Heart failure;
- A sharp decrease in hemoglobin levels (less than 50 g/l);
- Surgical intervention;
- Diseases of the musculoskeletal system;
- Pulmonary hypertension.
Therapy and prevention
To treat severe forms of thalassemia, blood transfusions are used to increase the number of red blood cells and restore oxygen transport, as well as chelation to remove the free form of iron that has not been incorporated into the “wrong” hemoglobin. In severe beta thalassemia, blood transfusions are needed frequently - every 3-4 weeks. However, now such therapy allows patients to lead a normal life and restores life expectancy. For mild forms of thalassemia, the indications are simpler - doctors advise maintaining a healthy lifestyle and moving more. Sports activities help maintain the mineralization of skeletal bones and prevent their deformation.
On a note
- Thalassemia is a severe hereditary disease that occurs due to mutations in the hemoglobin genes. It is characterized by weakness and dizziness, liver and heart disease, an increase in the size of the spleen, as well as bone deformation.
- There are two types of the disease - alpha and beta thalassemia. On average, in the world there are about five percent of carriers of mutations characteristic of both species, while in Russia it is only about one percent.
- Thalassemia is diagnosed using blood tests, ultrasound and genetic testing.
- To treat severe forms, frequent blood transfusions and removal of free forms of iron are used. For mild forms, maintaining a healthy lifestyle is sufficient.
Sources:
- Practical medicine, Hemoglobinopathies and thalassemic syndromes, 2015
- Subcellular Biochemistry, Hemoglobin: Structure, Function and Allostery, 2020
- Federal State Institution Federal Scientific and Clinical Center for Children's Health and Human Institutions of Roszdrav, Epidemiology of hemoglobinopathies in Moscow, 2008
- American Family Physician, Alpha and Beta Thalassemia, 2009
- NCBI, Thalassemia, 2021