Thalassemia is a genetic blood disease in which, due to gene mutations, an insufficient amount of hemoglobin is formed in the body and red blood cells are deformed.
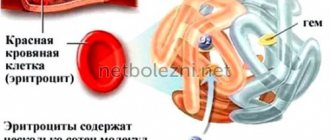
Hemoglobin is a specific protein that is found in red blood cells and is responsible for the transport of iron and oxygen to all organs and tissues. The main function of hemoglobin is respiratory and its deficiency leads to oxygen starvation. As a result, organs cannot fully perform their physiological functions.
There are “adult” hemoglobin (HbA) and fetal hemoglobin (HbF).
Fetal (fetal) hemoglobin is synthesized in the fetus 2 weeks after the formation of internal organs (from approximately the 12th week of pregnancy). After the birth of a child, HbF is up to 80% and is gradually replaced by “adult” HbA.
Normally, in a person, the fetal type of hemoglobin should remain no more than 1.5%. If there is a shift of numbers to the left or to the right, this always indicates a pathology (the functioning of the cardiovascular system, thyroid gland, pituitary gland may be disrupted, the presence of hematological abnormalities, leukemia, lymphogranulomatosis, intoxication).
A healthy mature hemoglobin molecule (HbA) consists of two pairs of chains - alpha and beta. Fetal hemoglobin (HbF) consists of chains - gamma and delta.
And thalassemia is the result of impaired synthesis of one or more hemoglobin chains. On the basis of which alpha thalassemia, beta thalassemia, beta delta thalassemia, etc. are distinguished.
The severity of one or another type of disease depends on the number of damaged areas:
- If one gene of the chain is changed, the disease (thalassemia minor) is mild and asymptomatic. But, at the same time, the person remains a carrier of this disease, which he can pass on to his children.
- Violation of the synthesis of 2 genes is manifested by mild microcytic anemia.
- The pathology of 3 genes is severe with pronounced symptoms.
- Damage to 4 genes at once (thalassemia major) is the rarest type of thalassemia, which is practically incompatible with life (sometimes the situation is solved by a bone marrow transplant).
Alpha thalassemia major is the most serious disorder that occurs during fetal development. It threatens complications for both the child and the woman.
Today, with timely intrauterine transfusion of red blood cells, it is possible to save the life of the fetus.
Beta thalassemia major (Cooley's anemia) is also a dangerous disorder that manifests itself in the first two years of a child's life.
Alpha and beta thalassemia minor disrupt the process of erythropoiesis (the development of new red blood cells), which leads to chronic anemia and impaired hemolysis.
Regardless of the type, the clinical course of thalassemia is similar, the differences depend on the severity of the pathological process.
What is thalassemia and why does it occur?
Thalassemia is a genetic blood disorder. An insufficient amount of hemoglobin, the protein responsible for transporting oxygen and carbon dioxide, is formed. It consists of four peptides. 90% of hemoglobin molecules contain two alpha globins and two beta globins (hemoglobin A or A1), 2.5% contain delta chains instead of beta chains (hemoglobin A2), and the rest are hemoglobin A that has aged during use in red blood cells ( hemoglobin A3). When a mutation occurs in the genes responsible for the synthesis of one of the globins, the composition of hemoglobin is disrupted. These changes lead to the death of red blood cells.
What are the prospects for Zinteglo?
Since Zinteglo is conditionally approved (without strict requirements, as in the case of other drugs), Bluebird is required to annually send updated clinical data to the regulator so that it renews the registration of the drug, based on the fact that the benefits outweigh the harm. As soon as the data is collected into a pool of the required volume, it will be possible to talk about a full-fledged marketing permit.
In the future, patients with a genotype characterized by a complete absence or critically low endogenous synthesis of beta-globin will be added to the spectrum of patients for whom Zynteglo gene therapy is indicated: for example, β0/β0, mutations IVS-I-110 or IVS-I-5. The effectiveness of treatment in such patients may be less clear: clinical trials of HGB-212 are testing this.
This year, Bluebird intends to submit a registration dossier for Zinteglo to the US Food and Drug Administration (FDA), with a launch expected in 2020.
Along the way, Bluebird is completing a test of Zynteglo in the gene therapy treatment of sickle cell anemia, since this pathology is caused by other mutations of the same beta-globin gene.
The estimated European population of patients with transfusion-dependent beta thalassemia is 11–13 thousand people, with half living in Italy. Commercial prospects in the United States are much more modest with a patient pool of 1,400–1,500 people. Taking into account the upcoming inclusion of sickle cell anemia, it is quite possible for Zinteglo to earn bestseller status, crossing the threshold of $1 billion in annual sales.
In terms of competition, luspatercept will see the light of day in the foreseeable future: the FDA will decide on its approval for transfusion-dependent beta thalassemia by early December. Luspatercept, being developed by Acceleron Pharma and Celgene, is an erythrocyte maturation activator. This soluble fusion protein, comprising a modified extracellular domain of activin receptor type IIB (ActRIIB) and the Fc domain of human immunoglobulin IgG1, targets specific ligands of the transforming growth factor beta (TGF-β) superfamily that regulate late-stage erythropoiesis. Luspatercept is thought to act as a ligand decoy that inhibits abnormal Smad2/3 signaling responsible for ineffective erythropoiesis.
Causes and risk factors for developing the disease
Thalassemia is a hereditary disease, meaning the mutation is passed from parent to child. If one of your relatives suffered from this pathology, the risk of developing the disease increases.
The second risk factor is ethnicity. Thalassemia is most common in Africa, Central Asia and the Mediterranean countries, where it was discovered (translated from Greek “thalassemia” is “sea anemia”). There are two main types of thalassemia: alpha and beta. In the first case, the mutation affects the genes responsible for the synthesis of alpha globins, in the second - beta.
Alpha globins are encoded by four genes. The severity of the disease will depend on the number of pathologically altered DNA sections:
- 1 altered gene – asymptomatic form. But with it, a person becomes a carrier of the disease and can pass it on to his children;
- 2 genes – mild course of the disease;
- 3 genes – severe course of the disease;
- 4 genes is a rare type of disease that is poorly compatible with life. Most fetuses die during fetal development, and newborn babies generally die soon after birth or require lifelong therapy. In some cases, they can be cured by bone marrow transplantation.
Beta globins are encoded by a single gene, which is localized on chromosome 11. If the defective gene is contained in only one chromosome of a pair, the disease is mild (thalassemia minor). Damage to both chromosomes results in a very serious disease known as thalassemia major or Cooley's disease (Cooley's anemia).
Frequency of occurrence
Thalassemia is inherited in an autosomal recessive manner. This means that it affects both boys and girls equally. The recessive nature suggests that children with thalassemia appear in those families where mom and dad are both carriers of mutations. Although they may not even realize that they are carriers, since the symptoms are sometimes invisible.
The average incidence is 1 in 100,000 people and may vary by region. Beta thalassemia is more common than alpha thalassemia.
Symptoms and signs of thalassemia
In most cases, thalassemia is detected at the stage of prenatal diagnosis. If necessary, treatment begins immediately, without waiting for symptoms to appear. If the disease is not detected by prenatal diagnosis, the following symptoms are expected:
- pallor or yellowness of the mucous membranes;
- slow growth;
- dark urine;
- abdominal enlargement;
- deformation of bones, especially the bones of the skull.
The time at which the first signs of thalassemia appear largely depends on the type of disease and the number of mutations. In some children, symptoms are registered soon after birth, in others - in the first two years of life.
Diagnosis of thalassemia
Symptoms of thalassemia are more or less characteristic. To make a final diagnosis, the doctor needs laboratory results. If thalassemia is suspected, a general blood test is required. It will show a reduced number of red blood cells that are small, light, different in shape and size. In addition to mature cells, the smear will contain many of their precursors - blasts. Additionally, other specific blood tests may be prescribed to determine the severity of the disorders (biochemical analysis, determination of iron-binding capacity of plasma or serum ferritin). Molecular tests (PCR) have also been developed to determine the presence of mutations.
Ultrasound is used to assess the condition of the liver and spleen, and radiography is used to identify bone tissue pathology.
In a child, thalassemia can be diagnosed during pregnancy. This study is especially recommended for parents who are sick or may be carriers of this disease. There are two diagnostic methods:
- chorionic villus biopsy - performed at the 11th week of pregnancy;
- amniocentesis (sampling of amniotic fluid) - prescribed at the 16th week.
Treatment of thalassemia
Determined by type and severity. For moderately severe symptoms, treatment is not prescribed. From time to time only blood transfusions are performed. This is mainly necessary after operations, childbirth, or to prevent possible complications. People with beta thalassemia require more frequent blood transfusions. To normalize excess iron levels, they are also prescribed specific drugs that bind and remove iron.
For severe and severe forms of the disease, there are two treatment options:
- frequent blood transfusions (every few weeks), which are combined with taking medications that remove excess iron from the body;
- Bone marrow transplant is the only method that can completely cure a person of thalassemia. Unfortunately, transplantation is not always successful.
Medicines for thalassemia are prescribed only to correct symptoms and complications. There is no drug therapy for the disease itself.
How much does Zinteglo cost?
PS Bluebird put the cost of Zinteglo at 1.575 million euros ($1.77 million). As expected, payment will be made in installments over five years in equal installments: first an advance, and then annual payments, but only if the effectiveness of treatment is recorded.
Bluebird has not yet determined the final cost of its gene therapy, but it will obviously be prohibitive. It is clear that in highly developed countries with medical insurance, none of the patients pays the entire amount out of their own pocket.
Yes, according to Bluebird's calculations, the "fair price" of Zinteglo, calculated taking into account the improvement in the patient's quality and life expectancy, is within $2.1 million, but the final cost is expected to be significantly less. This astronomical amount obviously comes from the total costs of the health care system for many years of chronic therapy for beta thalassemia. Thus, according to experts, one American patient with this disease costs somewhere between 200-300 thousand dollars annually.
It is proposed to pay 20% in advance in the first year, and the remaining amount to be paid in 20% installments annually for four years, provided that the effectiveness of treatment meets predetermined criteria, such as the disappearance of the need for blood transfusions or a significant reduction in the number of such procedures.
In other words, you will have to pay only based on the effectiveness of gene therapy, especially since there is still no indisputable data on the duration of its healing effect. The approach seems reasonable given the differences in intermediate outcomes among patients, although the overall effectiveness of Zynteglo is impressive.
According to industry experts, the cost of Zinteglo will be $1.2 million in the United States and $900 thousand (785 thousand euros) in Europe.
Complications of thalassemia
Possible complications of the disease:
- excess iron, which is part of hemoglobin. The accumulation of this element in the body leads to damage to the heart, liver, and endocrine system;
- susceptibility to infections. This is especially true for patients who have had their spleen removed;
- bone deformation associated with an increase in bone marrow volume. Most often, this process affects the bones of the skull, less often the limbs. They become thinner and break more often;
- splenomegaly is an enlargement of the spleen, where defective red blood cells mainly die. If the spleen is very enlarged, it is removed. This operation is called splenectomy;
- delayed growth and puberty;
- Heart diseases (chronic heart failure and arrhythmias) can develop in severe cases of the disease.