Гипертрофия миокарда: причины возникновения
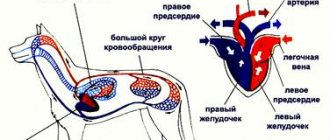
Механизм возникновения гипертрофии миокарда достаточно прост. Всем известно, что сердце выполняет «насосные» функции, перекачивая всю кровь в организме, доставляя кислород к каждому органу. Постоянные чрезмерные физические и эмоциональные нагрузки приводят к повышенной потребности всех органов в кислороде и, следовательно, к большому объему крови. Все это приводит к тому, что миокард не справляется с работой, теряет свою эластичность и увеличивается в своих размерах. Гипертрофия миокарда обычно возникает по следующим причинам: — артериальная гипертония; — приобретенные и врожденные пороки сердца; — нарушение кровоснабжения; — ожирение; — постоянные перегрузки на сердце; — генетическая предрасположенность… Необходимо еще отметить, что гипертрофией миокарда чаще всего болеют дети и данное заболевание является признаком врожденного порока сердца.
Гипертрофия левого желудочка неясной этиологии
С.В. Моисеев. Гипертрофия левого желудочка (ГЛЖ) часто встречается в практике кардиолога. Она может быть заподозрена на основании ЭКГ, однако более надежными методами оценки массы миокарда являются эхокардиография и особенно магнитно-резонансная томография (МРТ). При эхокардиографии критерием ГЛЖ считают увеличение индекса массы миокарда левого желудочка, соответственно, >115 г/м2 у мужчин и >95 г/м2 у женщин (с поправкой на площадь поверхности тела) или >50 г/м2,7 у мужчин и >47 г/м2,7 у женщин (с поправкой на рост) [1], а при МРТ – >85 г/м2 у мужчин и >81 г/м2 у женщин [2]. В зависимости от относительной толщины стенки (ОТС) левого желудочка [(2 × толщина задней стенки)/ конечный диастолический размер левого желудочка] выделяют концентрическую (ОТС≥0,43) и эксцентрическую (ОТС<0,43) ГЛЖ. Учитывая возможную неоднородность утолщения левого желудочка, в частности асимметричное увеличение толщины межжелудочковой перегородки или верхушки левого желудочка, при диагностике ГЛЖ следует учитывать увеличение толщины перегородки или стенки ≥12 мм.
У подавляющего большинства пациентов причиной ГЛЖ является перегрузка сердца давлением или объемом при артериальной гипертонии или пороках сердца, прежде всего аортальных, однако у части больных очевидные причины ГЛЖ отсутствуют. Распространенность необъяснимой ГЛЖ у взрослых в общей популяции составляет 0,02-0,23% [3]. Отсутствие явных причин гипертрофии миокарда обычно служит основанием для установления диагноза гипертрофической кардиомиопатии (ГКМП), хотя у 5-10% пациентов ГЛЖ неясной этиологии обусловлена другими генетическими и негенетическими заболеваниями, в том числе лизосомными болезнями накопления (болезни Фабри, Данона, Помпе), ATTR- и AL-амилоидозом, нейромышечными заболеваниями (атаксия Фридрейха), митохондриальными кардиомиопатиями и др. Своевременная диагностика некоторых из них, в частности болезней Фабри и Помпе, AL- и ATTR-амилоидоза, имеет важное практическое значение, учитывая возмож ность патогенетического лечения. При проведении дифференциального диагноза следует учитывать возраст пациента, выраженность ГЛЖ и клинических симптомов, наличие семейного анамнеза и различных экстракардиальных проявлений. Некоторые генетические заболевания, например, болезнь Фабри и ATTR-амилоидоз, иногда удается диагностировать только с помощью скрининговых исследований. Ниже будут рассмотрены наиболее распространенные причины ГЛЖ неясной этиологии.
Е.В. Привалова. ГКМП – это наследственное заболевание, которое передается по аутосомно-доминантному типу [4]. В соответствии с рекомендациями Евро пейского общества кардиологов 2014 г., ГКМП может быть диагностирована при наличии гипертрофии миокарда ≥15 мм по крайней мере в одном сегменте левого желудочка, которую нельзя объяснить другими причинами, а у родственников пациента с установленным диагнозом – при наличии гипертрофии миокарда ≥13 мм [3]. Гипертрофия миокарда при ГКМП может быть как асимметричной (рис. 1), так и симметричной. У 4060% пациентов с ГКМП определяются мутации генов, кодирующих белки саркомеров сердца, прежде всего тяжелой цепи бета-миозина (MYH7) и миозин-связывающего белка С (MYBPC3). Реже встречаются мутации генов, кодирующих тропонины I и T (TNNI3, TNNT2), α1-цепи тропомиозина (TPM1) и легкой цепи миозина 3 (MYL3). В целом у пациентов с мутациями генов саркомерных белков выше частота семейного анамнеза ГКМП и внезапной смерти и отмечаются более выраженные ГЛЖ и миокардиальный фиброз, чем у пациентов без мутаций [5]. При обследовании пациента важно оценить наличие обструкции выносящего тракта левого желудочка [6]. Критерием ее является градиент давления в выносящем тракте, который измеряют допплеровским методом, ≥30 мм рт. ст. в покое или после провокационных проб (проба Вальсальвы, физическая нагрузка). Гемодинамически значимым считают увеличение этого показателя ≥50 мм рт. ст. [3].
Рис. 1. Выраженнаяасимметричная гипертрофия межжелудочковой перегородки при ГКМП
В.Ю. Каплунова. Пациент К., 45 лет, обследован в клинике госпитальной терапии имени А.А. Остроумова в октябре 2021 г. Старший брат пациента внезапно умер в возрасте 54 лет. ГКМП была диагностирована у другого брата, умершего в возрасте 54 лет, и 29-летней дочери пациента. С 18-летнего возраста у больного выслушивался систолический шум по левому краю грудины при отсутствии клинических проявлений и хорошей переносимости физической нагрузки. В возрасте 27 лет появились одышка, сердцебиение, перебои в работе сердца, головокружение и дурнота, дискомфорт в области сердца при умеренной физической нагрузке. В возрасте 33 лет обнаружена асимметричная ГЛЖ (толщина межжелудочковой перегородки – 20 мм, задней стенки – 12 мм) с признаками обструкции выносящего тракта левого желудочка и градиентом давления в покое 45 мм рт. ст. Диагностирована обструктивная форма ГКМП. С 44-летнего возраста отмечает пароксизмы фибрилляции предсердий с последующим переходом аритмии в постоянную форму. При эхокардиографии было выявлено нарастание гипертрофии межжелудочковой перегородки до 31 мм и увеличение градиента давления до 94 мм рт. ст. в покое. В Научном центре сердечно-сосудистой хирургии им. А.Н. Баку ле ва выполнена миэктомия доступом из правого желудочка, которая привела к уменьшению степени обструкции выносящего тракта левого желудочка и диастолической дисфункции. При молекулярно-генетическом исследовании у пробанда и его дочери выявлена миссенс мутация в 22 экзоне гена, кодирующего тяжелую цепь β миозина (MYHT A870C).
Представленное наблюдение иллюстрирует типичные проявления и течение ГКМП: асимметричная гипертрофия межжелудочковой перегородки, которая была выявлена в молодом возрасте, медленно нарастала и длительное время не сопровождалась клиническими симптомами, в частности застойной сердечной недостаточностью, обструкция выносящего тракта левого желудочка с высоким градиентом давления в его полости, семейный анамнез (диагноз ГКМП и/или случаи внезапной сердечной смерти у близких родственников), мутация гена, кодирующего саркомерный белок, которая была обнаружена как у пробанда, так и его дочери. Лечение ГКМП обычно начинают с β-адреноблокаторов, не обладающих вазодилатирующей активностью, которые уменьшают градиент давления в полости левого желудочка и клинические симптомы. При их неэффективности могут быть использованы дизопирамид или верапамил. При выраженной гипертрофии межжелудочковой перегородки и высоком градиенте давления в левом желудочке может быть выполнена миэктомия, которая более чем в 90% случаев позволяет ликвидировать или значительно уменьшить обструкцию выносящего тракта, улучшить переносимость физической нагрузки и выживаемость [7].
Е.В. Привалова. Дифференциальная диагностика ГКМП с другими заболеваниями, сопровождающимися ГЛЖ, может представлять трудности, например, при наличии умеренной гипертрофии миокарда, особенно симметричной, не сопровождающейся обструкцией выносящего тракта левого желудочка, и при отсутствии семейного анамнеза. Мутации генов саркомерных белков определяются не у всех пациентов с ГКМП, а у части больных с необъяснимой ГЛЖ молекулярно-генетическое исследование не проводится по экономическим причинам. Следует учитывать, что асимметричная гипертрофия межжелудочковой перегородки, характерная для ГКМП, встречается и при других заболеваниях, в том числе вторичной гипертрофии миокарда при артериальной гипертонии. Причиной ГЛЖ могут быть не только перегрузка левого желудочка давлением, но и физические тренировки, хотя в крупном исследовании увеличение толщины стенки левого желудочка более 12 мм было выявлено всего у 1,7% из 947 спортсменов, занимающихся различными видами спорта, а толщина стенки не превышала 16 мм. Более частым эхокардиографическим признаком “сердца спортсмена» была дилатация полости левого желудочка, которая определялась в 38% случаев [8]. Важное значение для диагностики некоторых заболеваний, сопровождающихся ГЛЖ, имеет тщательный анализ клинической картины, позволяющий выявить те или иные экстракардиальные проявления болезни, которые отсутствуют при ГКМП. Проведение биопсии миокарда для подтверждения диагноза ГКМП не требуется, однако гистологическое исследование может быть обоснованным для исключения инфильтративных заболеваний, сопровождающихся утолщением стенки левого желудочка.
Е.А. Каровайкина. Болезнь Фабри – это редкое заболевание, которое характеризуется нарушением обмена гликофосфолипидов вследствие недостаточности или отсутствия лизосомного фермента – α-галактозидазы А [9]. Причиной дефицита этого фермента являются мутации гена GLA, расположенного на Х-хромосоме, поэтому типичные клинические проявления болезни Фабри наблюдаются чаще и более выражены у гемизиготных мужчин, однако они нередко встречаются и у гетерозиготных женщин. При классическом фенотипе болезни Фабри первые симптомы, в частности нейропатическая боль (эпизоды жгучей боли в кистях и стопах, возникающей при лихорадке, физической нагрузке, стрессе и быстрых изменениях температуры окружающей среды), ангиокератомы (поверхностные ангиомы, локализующиеся на передней брюшной стенке, в частности внутри или вокруг пупка, в паховой области, на ягодицах, верхних конечностях, губах; рис. 2), снижение или отсутствие потоотделения, желудочно-кишечные нарушения, появляются в детском или подростковом возрасте, а в возрасте 20-40 лет развивается поражение внутренних органов, в том числе сердца, почек (протеинурия и прогрессирующее снижение скорости клубочковой фильтрации) и центральной нервной системы (транзиторные ишемические атаки и инсульт). При атипичном “кардиальном» варианте заболевания ГЛЖ развивается в возрасте 40-50 лет и старше при отсутствии ранних симптомов. Результаты нескольких крупных скрининговых исследований свидетельствуют о том, что патогенные мутации гена GLA, ассоциирующиеся с развитием болезни Фабри, могут быть обнаружены у 0,5-1% пациентов с диагнозом ГКМП [10].
Рис. 2. Ангиокератомы в области пупка у пациента с болезнью Фабри
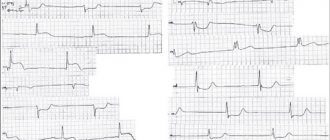
А.С. Моисеев. Иллюстрацией ГЛЖ, обусловленной болезнью Фабри, могут служить следующие два наблюдения. Пациент Л., 48 лет, был обследован в клинике им. Е.М. Тареева в октябре 2021 года. С 19 лет выраженная лимфедема нижних конечностей и снижение потоотделения. В возрасте 47 лет появились редкие боли за грудиной, не связанные с физической нагрузкой, а при эхокардиографии выявлено утолщение межжелудочковой перегородки и стенки левого желудочка до 14 мм при отсутствии дилатации камер сердца и нарушений систолической функции. Наблюдался у кардиолога с диагнозом ГКМП. Через 1 год при скрининге обнаружено снижение активности α-галактозидазы А в высушенных пятнах крови, увеличение уровня глобо триазилсфингозина (Lyso-GL3) до 117 нг/мл (в норме <1,8) и мутация в гене GLA (с.145C>G), которые позволили установить диагноз болезни Фабри. На ЭКГ отмечались признаки ГЛЖ (рис. 3). При МРТ сердца наблюдались увеличение индекса массы миокарда левого желудочка до 123 г/м2 и признаки интрамиокардиального фиброза. Кроме того, имелись симптомы поражения почек – снижение расчетной скорости клубочковой фильтрации до 62 мл/мин/1,73 м2 при отсутствии протеинурии.
Рис. 3. ЭКГ признаки гипертрофии миокарда у пациента с болезнью Фабри
Одновременно мы обследовали 67-летнюю мать пациента, у которой за 2 года до госпитализации в клинику также была выявлена необъяснимая ГЛЖ, сопровождавшаяся постоянной формой фибрилляции предсердий, частой желудочковой экстрасистолией и сердечной недостаточностью, в связи с чем принимала β-адреноблокаторы, мочегонные средства, дигоксин, антикоагулянты. При МРТ сердца определялись утолщение межжелудочковой перегородки (19 мм) и задней стенки левого желудочка (12 мм), увеличение индекса массы миокарда левого желудочка до 141 г/м2 и участки интрамиокардиального фиброза. При МРТ головного мозга выявлены многочисленные очаги в белом веществе, а при осмотре окулистом – воронковидная кератопатия, характерная для болезни Фабри, диагноз которой подтверждался результатами молекулярногенетического исследования (мутация с.145C>G), повышением уровня Lyso-GL3 до 23 нг/мл и снижением активности α-галактозидазы А. Пациент и его мать в течение 1,5 лет получают ферментозаместительную терапию.
Представленные наблюдения демонстрируют важность скрининга в диагностике болезни Фабри у пациентов с необъяснимой ГЛЖ, так как клинически заподозрить правильный диагноз было сложно в связи с отсутствием типичных ранних симптомов – нейропатической боли и ангиокератом. С целью диагностики болезни Фабри у мужчин необходимо определять активность α-галактозидазы А и/или уровень Lyso-GL3 в высушенных пятнах крови. У женщин активность фермента нередко остается нормальной или снижается незначительно, поэтому более информативным считают определение уровня Lyso-GL3. Для подтверждения диагноза проводят молекулярно-генетическое исследование с целью выявления патогенной мутации гена GLA (в Российской Федерации все эти исследования выполняются бесплатно).
Н.Р. Носова. При анализе клинической картины обращали на себя внимание позднее развитие ГЛЖ и наличие ее не только у пробанда, но и его матери. Как указано выше, поражение внутренних органов при болезни Фабри, в отличие от некоторых других наследственных болезней, у мужчин обычно отмечается в возрасте 30-40 лет, а у женщин – в более старшем возрасте. Наследование болезни Фабри сцеплено с Х-хромосомой, поэтому у матери пациента обычно наблюдаются те или иные проявления заболевания, хотя фенотип его может отличаться. У пробанда с 18-летнего возраста отмечались снижение потоотделения, которое встречается у большинства мужчин с болезнью Фабри, и лимфедема. По данным регистра Fabry Outcomes Survey (n=714), частота лимфедемы при этом заболевании составила 16% у мужчин и 6% у женщин [11]. У матери пробанда была выявлена вихревидная кератопатия (коричнево-золотистые отложения в роговице в виде волнообразных линий, исходящих из одной центральной точки), которая является одним из клинических критериев диагноза болезни Фабри. По нашим данным, частота вихревидной кератопатии у 69 взрослых пациентов с болезнью Фабри составила 65,2%, в том числе 56,4% у мужчин и 76,7% у женщин [12]. В отличие от ГКМП, гипертрофия миокарда как у пробанда, так и его матери была выражена умеренно и не сопровождалась обструкцией выносящего тракта левого желудочка. У взрослых пациентов с болезнью Фабри обычно наблюдается поражение не только сердца, но и почек (альбуминурия/протеинурия, снижение скорости клубочковой фильтрации) и головного мозга (очаговые изменения в белом веществе при МРТ, транзиторные ишемические атаки/инсульт).
Е.А. Каровайкина. При болезни Фабри проводят заместительную терапию рекомбинантными препаратами α-галактозидазы А (агалсидазой альфа в дозе 0,2 мг/кг или агалсидазой бета в дозе 1 мг/кг), которые вызывают регресс ГЛЖ или по крайней мере замедляют прогрессирование кардиомиопатии. По данным D. Ger main и соавт., у пациентов с болезнью Фабри, начавших лечение в более молодом возрасте (18-30 лет), средняя масса миокарда левого желудочка снижалась на 3,6 г в год, в то время как без лечения у мужчин того же возраста она увеличивалась на 9,5 г в год (р<0,0001) [13].
В.В. Рамеев. Причиной утолщения стенок сердца мо жет быть не только гипертрофия кардиомиоцитов, но и инфильтрация миокарда нерастворимым фибриллярным гликопротеидом – амилоидом. В настоящее время известно около 30 амилоидогенных белков, однако более 95% случаев амилоидоза сердца обусловлены ALамилоидозом или реже транстиретиновым (ATTR) амилоидозом. AL-амилоидоз развивается при отложении моноклональных иммуноглобулинов у больных лимфоплазмаклеточными дискразиями, в том числе множественной миеломой. ATTR-амилоид (мутантный и дикого типа) образуется из транстиретина, синтезируемого в основном печенью и выполняющего функции транспортного белка тироксина и витамина А. Причи ной развития ATTR-амилоидоза дикого типа (стар ческого) считают возрастное снижение активности ферментных систем гепатоцитов, что приводит к пре имущественнои секреции нестабильных мономерных форм транстиретина, которые легко агрегируют в тканях с образованием амилоида. В основе наследственного ATTR-амилоидоза лежат мутации в гене TTR, сопровождающиеся синтезом транстиретина, который не способен образовывать тетрамеры и обладает очень высокой амилоидогенностью.
А.С. Рамеева. Пациентка Б., 50 лет, впервые обследована в клинике им. Е.М. Тареева в январе 2015 года. В течение года беспокоили боли в области сердца и нарастающая сердечная недостаточность. При эхокардиографии выявлено утолщение межжелудочковой перегородки и задней стенки левого желудочка до 14 мм, нарушение диастолической функции по рестриктивному типу. При коронарографии обнаружен умеренный стеноз правой коронарной артерии (50%). При обследовании в российском кардиологическом научном центре в декабре 2014 года определялись небольшая протеинурия (0,14 г/л), снижение вольтажа зубцов ЭКГ (рис. 4), увеличение толщины межжелудочковой перегородки и задней стенки левого желудочка до 18 мм, дилатация левого предсердия при отсутствии дилатации левого желудочка и снижения фракции выброса, при МРТ сердца – диффузное неравномерное субэндокардиальное накопление контрастного вещества. Выс ка зано предположение об амилоидозе, диагноз которого был подтвержден при биопсии миокарда. При исследовании крови методом Freelite отмечено резкое увеличение концентрации свободных легких цепей лямбда типа до 1383 мг/л (в норме 5,7-26,3 мг/л), указывавшее на наличие AL-амилоидоза. При трепанобиопсии диагностирована множественная миелома (увеличение числа плазматических клеток до 20%). Пациентке проводилась терапия бортезомибом, мелфаланом и дексаметазоном, на фоне которой нормализовалась концентрация свободных легких цепей лямбда типа и достигнута компенсация сердечной недостаточности.
Рис. 4. Низкий вольтаж комплексов QRS в грудных отведенияхприамилоидозесердца
В.В. Рамеев. Таким образом, у пациентки был диагностирован AL-амилоидоз с поражением сердца в рамках множественной миеломы. Заподозрить амилоидоз сердца позволяли возраст пациентки, быстрое развитие тяжелой сердечной недостаточности, которая плохо поддавалась симптоматической терапии, эхокардиографические признаки рестриктивного поражения сердца (увеличение левого предсердия при отсутствии дилатации левого желудочка и снижения фракции выброса), симметричное утолщение межжелудочковой перегородки и стенки левого желудочка без обструкции выносящего тракта, снижение вольтажа зубцов комплекса QRS на ЭКГ. Последний признак отличает амилоидоз сердца от ГЛЖ, хотя истинное снижение амплитуды QRS (менее 5 мм в отведениях от конечностей и менее 10 мм в грудных отведениях) наблюдается только у половины больных AL-амилоидозом сердца [14]. Однако даже при отсутствии низкого вольтажа комплексов QRS следует учитывать возможное его несоответствие степени ГЛЖ при эхокардиографии. Важное диагностическое значение имеют результаты МРТ сердца, которая позволяет не только измерить массу миокарда левого желудочка, но и выявить диффузное накопление гадолиния в субэндокарде [15].
В представленном наблюдении диагноз был подтвержден при биопсии миокарда, хотя для гистологического исследования могут быть использованы и другие ткани, более доступные для биопсии, в том числе слизистая оболочка прямой или двенадцатиперстной кишки, подкожно-жировая клетчатка, почка. На наличие AL-амилоидоза указывала моноклональная секреция лямбда-цепей иммуноглобулинов, выявленная с помощью метода Freelite, а также снижение отношения каппа- и лямбда-цепей до 0,01 (для AL-амилоидоза характерна величина этого показателя <0,26 или >1,65). У 7-10% больных AL-амилоидоз развивается в рамках множественной миеломы, для исключения которой всем пациентам следует проводить биопсию костного мозга.
Данный случай демонстрирует возможность “изолированного» поражения сердца при AL-амилоидозе, хотя у большинства пациентов наблюдаются и другие проявления, в том числе протеинурия/нефротический синдром, увеличение печени и селезенки, макроглоссия, периорбитальная пурпура, диарея, невропатия и/или ортостатическая гипотензия. У пациентки имелась небольшая протеинурия, однако она могла быть связана с застоем крови по большому кругу кровообращения.
Современная химиотерапия, включающая в себя ингибитор протеасом бортезомиб, позволяет добиться полного или частичного гематологического ответа у значительной части больных AL-амилоидозом, предупредить отложение амилоида в других органах и прогрессирование сердечной недостаточности.
П.П. Тао. Пациент В., 65 лет, русский, был обследован в клинике им. Е.М. Тареева в декабре 2015 года. В течение трех лет отмечается нарастающее снижение болевой, температурной и тактильной чувствительности в области кистей и стоп по типу “перчаток» и “носков», а в течение одного года – прогрессирующая застойная сердечная недостаточность. При электромиографии выявлены грубые аксонально-демиелинизирующие нарушения, наиболее выраженные в малоберцовых нервах, а при эхокардиографии – картина рестриктивного поражения сердца: дилатация левого предсердия, утолщение стенок левого желудочка, зоны гипокинезии в межжелудочковой перегородке, уплотнение эндокарда, нормальная фракция выброса левого желудочка. Уровень мозгового натрийуретического пропептида был повышен в 30 раз по сравнению с верхней границей нормы. При коронарографии обнаружен стеноз передней межжелудочковой ветви (65%) и правой коронарной артерии (75%). Проведены чрескожная коронарная ангиопластика и стентирование правой коронарной артерии, однако после вмешательства сердечная недостаточность сохранялась, появилась ортостатическая артериальная гипотензия. По данным эхокардиографии, толщина межжелудочковой перегородки достигла 22 мм, определялись множественные очаги зернистости в миокарде, фракция выброса левого желудочка снизилась с 57% до 45%. При МРТ сердца с контрастированием гадолинием на фоне выраженного утолщения стенок левого желудочка отмечено циркулярное субэндокардиальное диффузное накопление контрастного вещества в миокарде левого и передней стенки правого желудочков (рис. 5). Проводилась повторная биопсия слизистой оболочки прямой кишки, однако амилоид выявить не удалось. При иммунохимическом исследовании исключена моноклональная секреция легких цепей иммуноглобулинов, характерная для AL-амилоидоза. При молекулярно-генетическом исследовании обнаружена мутация гена TTR (Val30Met), подтверждающая диагноз наследственного ATTR-амилоидоза. При сцинтиграфии миокарда с 99mTcPYP выявлено накопление радиоизотопного препарата 2 степени, в связи с чем от биопсии миокарда было решено воздержаться. С целью восстановления тетрамерной структуры мутантного транстиретина пациент в течение 2 лет получает тафамидис 20 мг/сут. Переносимость лечения удовлетворительная, существенного прогрессирования амилоидоза не произошло.
Рис. 5. Симметричное утолщение стенки сердца и накопление гадолиния в субэндокарде при МРТ у пациента с ATTRамилоидозом
В.В. Рамеев. Как и в предыдущем наблюдении, заподозрить амилоидоз у 65-летнего пациента позволяло типичное рестриктивное поражение левого желудочка, характеризующееся развитием тяжелой сердечной недостаточности при отсутствии дилатации и существенного снижения фракции выброса левого желудочка. При эхокардиографии определялись множественные очаги зернистости, которые нередко выявляют у пациентов с амилоидной инфильтрацией миокарда, а при МРТ сердца – диффузное накопление гадолиния в субэндокарде. В пользу системного амилоидоза свидетельствовала и периферическая полиневропатия, которая встречается как при AL-, так и семейном ATTR-амилоидозе и может предшествовать поражению сердца. В большинстве случаев развивается неуклонно прогрессирующая, симметричная дистальная невропатия, начинающаяся с сенсорных расстройств, в первую очередь болевой и температурной чувствительности, с последующим присоединением нарушений вибрационной и позиционной чувствительности и двигательных нарушений. Ранними симптомами невропатии бывают парестезии или мучительные дизестезии. Часто встречается синдром запястного канала, проявляющийся болями и парестезиями в I-III пальцах кисти с постепенной атрофией мышц тенара и обусловленный сдавлением срединного нерва в запястном канале амилоидом, откладывающимся в связках запястья [16].
Учитывая отсутствие моноклональной секреции легких цепей иммуноглобулинов и наличие мутации гена TTR, был установлен диагноз наследственного ATTRамилоидоза [17]. Диагноз амилоидоза должен быть подтвержден при гистологическом исследовании, однако результаты повторной биопсии прямой кишки оказались отрицательными. Тем не менее, наличие ATTRамилоидоза не вызывало сомнения с учетом типичной клинической картины и результатов молекулярно-генетического исследования. Кроме того, при сцинтиграфии было выявлено накопление 99mTcPYP в миокарде 2 степени (т.е. умеренное накопление, соответствующее таковому в костной ткани). В 2021 году в многоцентровом исследовании было показано, что накопление в миокарде 99mTcPYP 2-3 степени (рис. 6) при отсутствии моноклональной гаммапатии обладает 100% специфичностью в диагностике ATTR-амилоидоза сердца и фактически позволяет отказаться от биопсии миокарда [18]. Более того, сцинтиграфия с 99mTcPYP дает возможность дифференцировать ATTR-амилоидоз от AL-амилоидоза, при котором накопление радиоактивного препарата в миокарде отсутствует или не превышает 1 степени.
Рис. 6. Накопление 99mTcPYPв миокарде 3 степени при ATTR-амилоидозе
С середины 90-х годов ХХ века для лечения ATTRамилоидоза применяли трансплантацию печени, позволяющую восстановить синтез нормального транс тиретина. В последние годы более перспективной тактикой лечения считают медикаментозную стабилизацию тетрамерной структуры транстиретина и предотвращение образования амилоидогенных мономеров белка. Первый такой препарат – тафамидис – уже применяется в Европе и в Российской Федерации.
С.В. Моисеев. Представленные наблюдения иллюстрируют широкий спектр причин ГЛЖ неясной этиологии, которые включают в себя не только истинную гипертрофию миокарда, но и некоторые инфильтративные заболевания, такие как системный амилоидоз, имитирующие ГКМП. Разработать четкий алгоритм дифференциальной диагностики ГКМП достаточно сложно, учитывая вариабельность течения заболеваний, сопровождающихся ГЛЖ. Например, некоторые тяжелые генетические заболевания, такие как болезнь Помпе (гликогеноз II типа, связанный с дефицитом фермента кислой α-глюкозидазы в лизосомах), могут проявиться в возрасте 40-50 лет и старше, в то время как при системных заболеваниях, в том числе болезни Фабри или амилоидозе, экстракардиальные симптомы иногда отсутствуют. Ключевую роль в диагностике наследственных заболеваний, таких как ГКМП, ATTRамилоидоз, болезни Фабри, Помпе, Данона и др., играют изучение семейного анамнеза и молекулярногенетическое исследование.
Гипертрофия миокарда: лечение
Целью лечения гипертрофии миокарда является уменьшение размеров желудочка сердца. Для решения этой задачи необходим комплексный подход, который выражается в изменении образа жизни, отказе от вредных привычек: — исключите по максимуму из своего рациона пищу с высоким содержанием животных жиров; — откажитесь от курения и злоупотребления алкоголем; — ведите активный образ жизни (как на работе, так и на отдыхе); — займитесь закалкой организма (баня и контрастный душ — прекрасные средства); Очень эффективна комплексная терапия с применением Трансфер фактор Кардио — иммунного препарата на основе трансферфакторных иммунных молекул, которые, попадая в организм, выполняют 3 функции: — устраняют нарушения в структуре нашей ДНК, что и является настоящей причиной заболеваний; — усиливают нашу иммунную систему в отношении заболеваний сердца и сосудов; — выполняют роль носителей «иммунной памяти», записывая всю информацию о чужеродных элементах с которыми пришлось сталкиваться нашему организму и методы их нейтрализации. Эти свойства данного иммуномодулятора делают его самым эффективным в мире, так что его помощь требуется не только для комплексного лечения гипертрофии миокарда, но и при любых заболеваниях сердца и сосудов. Гипертрофия миокарда, лечение которой мы рассмотрели выше, на самом деле не является «приговором». При соответствующем образе жизни, любой человек вполне может избавиться он этого заболевания и не вспоминать о нем, но прием иммуномодулятора просто необходим.
Симптомы
Как правило, данная патология обнаруживается случайно, в процессе проведения ЭКГ или Эхо-КГ. Специалисты выделяют такие основные признаки гипертрофии миокарда:
- стенокардию;
- мерцательную аритмию;
- повышенное артериальное давление;
- общую слабость, плохое самочувствие.
Развитие стенокардии происходит в результате сжатия сосудов, обеспечивающих необходимым питанием сердечную мышцу. В конечном итоге происходит увеличение мышцы в размерах. Она начинает потреблять больше кислорода в комплексе с питательными веществами. У пациентов, страдающих от гипертрофии миокарда сердца, также может возникать состояние, в процессе которого замирает сердце на несколько мгновений (не бьется вообще). В этом случае человек теряет сознание.
Получить бесплатную консультацию Консультация ни к чему Вас не обязывает
Диагностика
Как правило, заболевание выявляется при ультразвуковом исследовании. МРТ сердца считается наиболее простым, информативным методом. В определенных случаях заболевание диагностируют при помощи ЭКГ. К дополнительным исследованиям относят: вентрикулографию, коронарографию, а также радиоизотопное исследование.
Профилактика
К числу эффективных профилактических действий по предотвращению развития заболевания относят:
- изменение образа жизни: отказ от курения, употребления алкогольных напитков;
- борьбу с факторами риска: нормализацию массы тела, артериального давления;
- контроль препаратами иперлипидемии и гипертонии (если при коррекции образа жизни отсутствует результат).
В случае контроля препаратами удается поддерживать нормальный уровень сахара, а также контролировать остальные факторы риска, возникающие при сахарном диабете. Соблюдение таких профилактических мер позволяет избегать развития гипертрофии миокарда сердца.