Coagulopathy, the symptoms of which are caused by serious disturbances in both the blood coagulation system and the anticoagulant mechanisms, is characterized by heavy spontaneous bleeding. As a rule, in this case they manifest themselves in the form of hematomas. This pathological process can be congenital, then it develops as a result of a lack of necessary blood-clotting components contained in the blood plasma.
Sometimes this condition can develop during the life of a sick person. In this case, the components necessary for blood clotting are usually present in sufficient quantities, but their quality is not up to standard. However, in both cases, this pathology is caused by certain genetic disorders. Most often, the first symptoms of coagulopathy appear in early childhood or in women during pregnancy.
Why does coagulopathy develop?
Before considering the symptoms and treatment of this pathology, it is necessary to determine the causes of its development. First of all, let's figure out how the process of blood clotting occurs normally. In the absence of serious violations, it takes place in three stages:
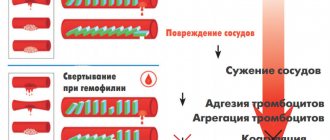
- With traumatic damage to the vascular wall of small blood vessels, a primary platelet clot is formed within the first five minutes.
- Over the next 15 minutes, with the help of fibrin, which is formed as a result of blood clotting, the primary thrombus is strengthened.
- The third stage is due to the need to free the lumen of the vessel from an unnecessary thrombus, which leads to its resorption.
The pathogenesis of coagulopathy is associated with any disorders that occur during one of the stages described above.
Relevance
Newborns and children in the first months of life are at particular risk for the development of hemorrhagic disorders. Of particular concern in this case is the potentially high level of serious complications that arise against the background of pathological changes in hemostasis [1–6]. The absence of specific manifestations of hemorrhagic syndrome, the variety of etiological factors and certain difficulties in conducting and interpreting the results of hemostasis studies in patients of this age category often determine a formulaic approach to the choice of therapy. This often leads to insufficient effectiveness of treatment and is accompanied by polypharmacy. Of particular concern is the low level of awareness among practicing pediatricians about the causes, clinical features, as well as modern diagnostic and treatment options for one of the most common causes of hemorrhagic syndrome in infants—vitamin K-deficiency coagulopathy (VKD) [7] . All this prompted the authors to prepare a publication in which, based on an analysis of modern literature data and their own clinical studies, the clinical, anamnestic and laboratory features of VKDK in infants are highlighted, and a diagnostic algorithm and treatment tactics are presented.
Main types of pathology
This disease can be of two main types:
- A congenital pathology, it is caused by a decrease in the quantitative or qualitative composition of homeostasis components. This is the name of the system responsible for the blood clotting process. There are several forms of this pathology. Each of them is caused by a deficiency of one component, which is reflected in their classification. The most common hemophilias are marked with the letters of the Latin alphabet: A, B and C. There are other forms of this pathology, but they are rarely registered. An example would be afibrinogenemia, which is characteristic of both men and women.
- Acquired pathological processes are a complication of any disease. These may be tumor processes, some infectious diseases or liver pathologies. The disease can also be caused by taking medications unsupervised by a doctor. Also, acquired coagulopathy can be a consequence of massive blood loss. The reason in this case is the lack of protein fractions and elements of the patient’s blood.
Let's look at the most common forms of this disease.
Congenital forms of pathology
Let us determine right away that almost all forms of congenital coagulopathy are caused by the absence or deficiency of only one factor. All of them have a common name - hemophilia. These pathological processes are hereditary in nature, most often associated with the gender of the sick person. All of them are caused by a lack of thromboplastin. Depending on which factor of this process suffers, the following types are distinguished:
- hemophilia A is caused by a lack of antihemophilic globulin;
- group B – imbalance of the Christmas factor;
- group C develops with a deficiency of the precursor thromboplastin, which is called “factor No. 9”.
The clinical picture of these pathologies does not differ significantly. Hemophilia A and B affect only men, and the disease is transmitted by women, that is, it is associated with the X chromosome. Group C coagulopathy is not associated with X-chromosomal inheritance, so both men and women can suffer from this disease.
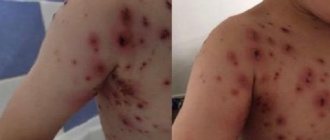
The symptoms of the disease are quite characteristic. These are prolonged bleeding that appear even with a minor injury or bruise. They have the appearance of edema, formed as a result of the sweating of blood into the surrounding tissues; petechiae do not form in this case. This is a characteristic feature in differential diagnosis. Almost spontaneous hemorrhages in the joint area are often diagnosed. Most often they affect the knee joints. In addition, clinical manifestations of hematuria and bleeding of the mucous membranes are common. Other types of congenital coagulopathy are very rare.
Hereditary coagulopathies: hemophilia and von Willebrand disease
The human blood coagulation system is a multicomponent and extremely complex mechanism that plays a critical role in protecting the entire body.
This mechanism is represented by three links: vascular, platelet and plasma coagulation. Diseases associated with the inability of blood coagulation factors to ensure the normal process of clot formation constitute a large group of coagulopathies. This group of diseases is represented by hereditary coagulopathies and many acquired forms of coagulopathies, which are the result of other diseases: cirrhosis and liver cancer, autoimmune (systemic lupus erythematosus, immune thrombocytopenic purpura, hemorrhagic vasculitis, etc.), infectious diseases, toxic effects of drugs and poisons, hereditary metabolic diseases.
Hereditary diseases of hemostasis
Hemophilia and von Willebrand disease are the most common hereditary diseases of the plasma hemostasis system. Von Willebrand factor and factor VIII in the blood plasma are presented in the form of a molecular complex, while von Willebrand factor plays a protective role for factor VIII, protecting it from destruction by protein C.
This is why, in the absence of von Willebrand factor, factor VIII levels can be significantly reduced. Thus, despite the differences between these diseases, they exhibit a certain molecular biological synergy. In this regard, it is advisable for practicing physicians to consider two diseases that account for more than 90% of hereditary coagulopathies caused by a deficiency or functional failure of fiery blood coagulation factors - VIII, IX (hemophilia A or B, respectively), or von Willebrand factor (von Willebrand disease).
Factor VII deficiency is also extremely rare. Hypoproconvertinemia manifests itself as a hemorrhagic syndrome, depending on the severity of factor VII deficiency. In severe cases, hemarthrosis, hematomas are observed, and in women, menorrhagia.
Both occur and are diagnosed infrequently
In general, hereditary coagulopathies are uncommon and even less commonly diagnosed. There are two main reasons for this: low prevalence, often associated with a subclinical course of the disease (in von Willebrand disease) and an insufficiently developed system for laboratory diagnosis of hemostasis disorders in most regions of our country. The number of patients with hemophilia in Russia is just over 7.5 thousand people, and with von Willebrand disease there should be about 16 thousand. The exact figure has not been established. Rare hereditary forms of coagulopathies - hypoproconvertinemia (factor VII deficiency), hypo- and afibrinogenemia, deficiency of factors XII, XIII, XI, V are extremely rare.
Hereditary coagulopathies are incurable, but the possibilities of modern drug therapy make it possible to provide patients with a duration and quality of life comparable to that in the general population.
Hemophilia, von Willebrand disease and hypoproconvertinemia belong to the group of social diseases, and without government support, people suffering from hereditary forms of coagulopathies are doomed. This is primarily due to the provision of expensive drugs for blood clotting factors, which patients receive for life at home. Treatment of these patients throughout the world is carried out in specialized hemophilia centers that maintain a medical register of patients with hereditary coagulopathies, and is based on national standards and treatment protocols. The medical register allows you to track the dynamics of a patient’s health over many years and adjust treatment.
"Calling card" of hemophilia
The clinical course of hemophilia and von Willebrand disease is different, but may have some similarities. The presence of hemarthrosis is a “calling card” of hemophilia, but it should be remembered that hemarthrosis can also be observed in von Willebrand disease (the so-called type III), and sometimes with severe hypoproconvertinemia (factor VII deficiency). Hemophilia is a disease caused by various types of mutations in the factor 8 or 9 gene, which results in a hereditary deficiency of clotting factor VIII (hemophilia A) or IX (hemophilia B).
These genes are localized on the long arm of the X chromosome and are inherited in a recessive manner, transmitted through women only to male children. In the population, the level of factors VIII and IX varies from 100 ± 50%, but in women - conductors of hemophilia - it may be lower than normal due to the functional inferiority of one of the two genes. The severity of hemophilia depends on the level of factor VIII or IX activity. In severe hemophilia, factor VIII (or IX) is absent or shows residual activity (less than 2%). In this case, the disease usually appears from early childhood.
Bleeding is typical when the integrity of the mucous membranes and skin, hematomas (bleeding from the umbilical cord, cephalohematoma, ecchymosis) are violated. When the child begins to walk, the first hemorrhages in the joints appear. In the moderate form (from 2 to 5%), damage to the musculoskeletal system is also noted, and in the mild form (more than 5%), the disease usually manifests itself during injuries and surgical operations, which may be accompanied by severe bleeding due to the rapid depletion of endogenous factor VIII or IX . Late diagnosis of the disease can lead to tragic consequences.
Today such a scenario can be avoided
The occurrence of primary hemarthrosis, even after its visible elimination, causes invisible changes in the cartilage tissue of the joint, which can be recorded on an NMR tomogram. Disintegrated red blood cells form an environment for the occurrence of a secondary aseptic inflammatory process, and the resulting hemosiderin is deposited in the cartilage tissue of the articular surfaces.
Subsequent hemorrhages expand the affected area, lead to the development of chronic synovitis, contributing to the occurrence of “spontaneous” hemorrhages that accompany the patient throughout his life, ultimately leading to ankylosis and muscle atrophy. Before reaching adulthood, such patients previously became disabled with multiple lesions of the musculoskeletal system. During the life of these patients, as a rule, there are massive hematomas, gastrointestinal bleeding and other various hemorrhages that pose a threat to the patient’s life or lead to his death. Modern drug therapy makes it possible to avoid such a scenario for the development of the disease.
It should be noted that throughout the life of a hemophilia patient, the level of the factor does not change, so replacement therapy remains the only non-alternative solution today. The constant presence of factor VIII at least 5% creates conditions for normalizing the rate of clot growth, and clinically determines the absence of “spontaneous” hemorrhages.
It is known that the half-life of factor VIII and IX inactivation (T?) is short. On average, it is 12 and 24 hours, respectively, but it can vary significantly for each patient. In hemophilia A, this range for factor VIII ranges from 7 to 20 hours. This indicator is important to consider when correcting the hemostatic system in patients with hemophilia, especially if treatment is carried out for a long time.
Studies conducted in our center using the method of spatial dynamics of blood coagulation make it possible to assess the level of “sufficiency” of replacement therapy in patients with coagulopathies. Compensation for the level of the missing coagulation factor should be in the “corridor” between hypo- and hypercoagulation and maintained in a given range throughout the patient’s life. Such replacement therapy to prevent hemorrhagic episodes should be determined individually. This selection, with the simultaneous use of classical methods for determining blood coagulation factors, allows you to set optimal therapy parameters calculated for a specific patient and determine the amount of drug required for treatment.
The most vulnerable link in the treatment of these patients is determining the need for prescribing blood clotting factors. All existing blood clotting factors are administered intravenously, i.e. their bioavailability is 100%, however, the catabolism of these complex protein structures depends on many individual parameters of the entire hemostatic system.
Today, preventive replacement therapy for patients with hereditary coagulopathies is selected empirically, often guided only by the initial diagnosis and the visible clinical result, which is assessed very subjectively. The same applies to single therapeutic doses aimed at stopping a hemorrhagic episode. For widespread clinical practice, such tests seem difficult, at least for today.
Treatment on demand and according to the situation
In order to formalize the standards of treatment for patients with hemophilia, a Protocol for the management of patients with hemophilia was developed and approved in 2005. This event fundamentally changed the quality of life of patients.
Replacement therapy with blood clotting factors can be prescribed both for the purpose of preventing hemorrhages - the so-called. “preventive treatment”: for patients with hemophilia A - 25 IU/kg body weight 3 times a week; for patients with hemophilia B - 25 IU/kg - 2 times a week, and to stop bleeding (symptomatic hemostatic therapy - “treatment on demand” - from 20 to 50 IU/kg and further depending on the clinical situation). These two treatment models can be alternated throughout the patient's life and are the “cornerstone” of the protocol.
Treatment of the disease can be carried out not only for the purpose of preventing hemorrhages. The protocol for the management of patients with hemophilia provides a model for the treatment of existing bleeding or hemorrhage. In this case, the peak level of factor VIII or IX 30 minutes after injection should be 40-100%, depending on the clinical situation.
Injections of the drug are repeated every 12 hours for hemophilia A and every 24 hours for hemophilia B at a dose of ? from initial to disappearance of symptoms of hemorrhage. Our experience shows that this treatment regimen is acceptable for patients with isolated hemorrhagic complications. As with many other diseases, in hemophilia it is easier to prevent hemorrhages rather than treat their complications.
General recommendation document
Nevertheless, attending hematologists should remember that this document is of a general recommendation nature, and the approach to treating each patient should be individual, sometimes taking into account the psycho-emotional profile of the patient and even his family members.
Hereditary coagulopathies are incurable, but the possibilities of modern drug therapy make it possible to provide patients with a duration and quality of life comparable to that in the general population.
Intravenous administration of the drug is carried out by the patient himself or his relatives after completing a specialized training program, and the patient is monitored by a hemophilia center or a hematologist.
This treatment is called "home treatment" and is used throughout the world. In our country, all patients with hemophilia are currently undergoing “home treatment”.
It has also been established that the phenotype of the disease may differ from its genotype. Not all patients with the same level of factor VIII or IX require the same treatment. 10-15% of patients with severe hemophilia A do not need preventive treatment at all, i.e. in the constant administration of drugs, however, all patients with hemophilia, without exception, require lifelong provision of blood clotting factors VIII or IX. This also applies to other hereditary forms of coagulopathies - von Willebrand disease and hypoproconvertinemia (factor VII deficiency).
Modern antihemophilic drugs of blood coagulation factors VIII and IX, from the point of view of etiotropic therapy aimed at achieving a clinical result, do not differ significantly from each other. All of them compensate for the level of the missing clotting factor to the same extent. Their activity is expressed in standard international units (IU). Conventionally, they can be classified into blood coagulation factors VIII containing von Willebrand factor and those not containing. The quantity and quality of von Willebrand factor may vary significantly between them. There are drugs in which the content of von Willebrand factor is increased. There is an isolated von Willebrand factor that does not contain factor VIII.
Recombinant clotting factors
Factor VIII plasma preparations that do not contain von Willebrand factor, the amount of which is not changed in patients with hemophilia, include drugs that have undergone affinity chromatography, as well as genetically engineered (recombinant) blood coagulation factors VIII (INN: octocog alpha). The Octocog alpha group of drugs is represented by three generations of drugs (classification is arbitrary): the first, containing added albumin, necessary to stabilize the factor VIII molecule, the second, containing traces of human albumin, and the third, free from the presence of albumin.
The basis of effective therapy for hereditary coagulopathies is: early diagnosis, selection of the correct treatment model and complete, continuous provision of patients with blood coagulation factors VIII or IX. If the amount of the drug is not enough for treatment, the disease begins to progress steadily and can negate all previously made efforts.
There is a recombinant factor VIII with a modified molecule, i.e. removed glycoprotein fragment (moroctocog alpha). The experience of its use is not as extensive as that of previous drugs. For patients with hemophilia B, coagulation factor IX preparations can be used - recombinant (nanokog alpha) and obtained from donor plasma, as well as prothrombin complex preparations (PPSB).
In patients with von Willebrand disease, it is more appropriate to use drugs containing a physiological or greater physiological ratio of von Willebrand factor to factor VIII. These drugs were developed specifically for the treatment of patients with von Willebrand disease, and the contents of the latter are indicated on the bottle label.
Almost absolute virus safety
The accumulated experience in the use of recombinant and plasma drugs indicates almost absolute viral safety against the viruses HIV-1, HIV-2, hepatitis B and C. Over the past 20 years, not a single case of infection of a recipient has been reliably recorded, however, discussions based on the “potential threat" continues.
Complications in hemophilia, as a consequence of treatment, are also inevitable. The most dangerous, but rare in Russia (about 3-5% of patients with hemophilia A) is the emergence of resistance to therapy, caused by the formation of immunoglobulins, often class G, the so-called. autoantibodies that selectively block the procoagulant activity of the factor VIII molecule. In hemophilia B, the formation of antibodies is extremely rare (less than 1-2%).
Cases of the formation of antibodies to von Willebrand factor have been described. Antibodies to factor VII are practically not formed. It is important for practicing hematologists to know that antigenic stimulation (drug administration) can stimulate the immune response and cause complete tolerance to replacement therapy. An inhibitor should be suspected if previously effective therapy is ineffective. The etiology and pathogenesis of this phenomenon remain unclear.
Diagnostics in regions is not available
Therapy and its monitoring in patients with hemophilia complicated by an inhibitor poses significant difficulties even for experienced specialists and laboratory workers. Fundamentally, there are two types of therapy: activation of the hemostatic system through shunting (bypass) pathways. For this, recombinant activated factor VII or prothrombin complex (activated) is used - blood coagulation factors II, VII, IX, X in combination. The second treatment option is immune tolerance induction, usually the Bonn Protocol. In hospital practice, plasmapheresis or immunoadsorption with protein A can be used, but this therapy leads to a temporary decrease in the titer of the inhibitor and is currently rarely used. Treatment of inhibitory hemophilia patients represents a serious medical and social problem.
In von Willebrand disease, characterized by decreased activity of von Willebrand factor, ristocetin-induced platelet aggregation, often with decreased levels of factor VIII and factor VIII antigen, it is important to determine whether there is a deficiency in the production of von Willebrand factor (quantitative deficiency - type I) or the structure of the factor molecule synthesized defective (qualitative deficiency - type II).
Diagnosis of the disease is quite complex and often unavailable in the regions, because requires complex coagulation studies. Important symptoms of the disease are prolonged nosebleeds, in women, prolonged mensis, which can lead to iron deficiency anemia, prolonged bleeding after tooth extraction or minor surgical interventions, and the manifestation of the same symptoms in close relatives (father, mother, brother, sister). Patients with such symptoms should be examined in specialized centers.
Extremely rare disease
An extremely rare disease is hereditary factor VII deficiency. With a pronounced deficiency of this factor, severe hemorrhagic syndrome is observed, accompanied by hematomas, hemarthrosis, and in women - life-threatening menorrhagia.
To treat patients with hypoproconvertinemia, blood coagulation factor VII (plasma) can be used at a dose of 30-40 IU/kg every 8-10 hours. In its absence, activated eptacog alfa at a dose of 20-40 mcg/kg every 2-4 hours until bleeding stops completely.
The basis for effective therapy for hereditary coagulopathies is: early diagnosis, selection of the correct treatment model and complete, continuous provision of patients with blood coagulation factors VIII or IX. If the amount of the drug is not enough for treatment, the disease begins to progress steadily and can negate all previously made efforts.
Monitoring of patients with hereditary coagulopathies is carried out throughout their lives in specialized centers, often together with doctors of other specialties if concomitant diseases arise. The organization of such centers in Russia is the organization of “treatment technology” for patients suffering from disorders of the blood coagulation system.
Acquired forms of the disease
There are quite a large number of conditions that manifest themselves as coagulopathic bleeding. They do not depend on the gender and age of patients and are most often not associated with genetic predisposition. Let's look at some of them.
Sometimes this condition may be associated with an insufficient amount of prothrombin produced. It is formed in the intestinal lumen in the presence of vitamin K, gastric juice and bile. In case of intestinal infections, liver damage or vitamin deficiencies caused by a lack of an essential vitamin, the production of prothrombin decreases or the process of its absorption is disrupted.
Consumptive coagulopathy can occur quite often. It is associated with the accumulation of fibrin in the bloodstream, which causes a decrease in fibrinogen. All this leads to areas of platelet accumulation with the development of the so-called symptom of thrombocytopenia. Which ultimately results in increased tissue bleeding. The cause may be septic conditions, massive trauma or pathological delivery.
The development of coagulopathies can also be caused by taking certain medications. Most often, this condition develops when taking certain direct (for example, Heparin) or indirect (Sinkumar, Pelentan) anticoagulants. It should be noted that bleeding can occur not only during an overdose, but also when using normal therapeutic doses of these drugs. This must be remembered during pregnancy. Clinically, this condition manifests itself in the form of increasing bleeding.
Clinical manifestations and diagnosis of vitamin K deficiency
In cases where the above etiological factors are not promptly addressed, vitamin K deficiency increases, which ultimately leads to the development of hemorrhagic syndrome in the child. As a rule, the clinical manifestation of VKDK is noted after 3–4 weeks. life of a child, most often this occurs at 1.5–2 months of age. At the same time, it is very important to remember that the maximum effectiveness of treatment measures for VKDK is achieved in cases where therapy begins with minimal hemorrhagic manifestations [11, 17]. In this regard, during an objective examination, it is necessary to pay attention to even the most minor hemorrhagic symptoms, which will become the basis for clarifying their causes. At this age, hemorrhagic syndrome is characterized by low specificity, and the maximum effectiveness of treatment is achieved only with an etiopathogenic approach, so searching for the cause becomes very important. Considering that hemorrhagic syndrome in infants can develop catastrophically quickly, it is necessary, without wasting time, to collect anamnesis and urgently (according to cito!
) perform a clinical blood test with the study of platelets and platelet indices, a coagulogram, a biochemical blood test, determine the blood group and Rh factor, and also conduct neurosonography due to the risk of intracranial hemorrhage (Fig. 1).
When collecting a family history, special attention should be paid to the presence of diseases in the child’s closest relatives that are accompanied by hemorrhagic syndrome (thrombocytopenia, thrombocytopathy, coagulopathy, thrombophilia). It is also very important to find out whether the child is prescribed medications that may cause hemorrhagic syndrome. In addition, in cases where the child is breastfed, it is necessary to clarify which medications are used by the mother. Anamnestic risk factors such as prematurity, long-term antibiotic therapy, and parenteral nutrition without vitamin supplementation must also be taken into account. Separately, it is necessary to clarify whether the child has hereditary metabolic disorders, congenital infections, malformations of the liver, gall bladder, intestines, as well as severe acquired diseases of the hepatobiliary and intestinal systems (Fig. 1).
When examining a child with hemorrhagic syndrome, regardless of the nature and severity of clinical manifestations, it is necessary to evaluate the level of consciousness, the color of the skin and visible mucous membranes, the color of feces and urine, the condition of the peripheral lymph nodes, liver, and spleen. In this case, the presence of icterus of the mucous membranes and skin with simultaneous lightening of stool and darkening of urine suggests damage to the hepatobiliary tract with the development of cholestasis. In these cases, first of all, it is necessary to think that the hemorrhagic syndrome is probably caused by acquired disorders of secondary hemostasis, since the synthesis of plasma factors of the coagulation system occurs in the liver.
A separate analysis requires a detailed description of the clinical manifestations of hemorrhagic syndrome. Thus, in the case of increased bleeding and hemorrhage, it is necessary not only to indicate the location (mucous membranes, umbilical wound, gastrointestinal tract, injection site, etc.), but also to note the time when the indicated symptoms first appeared, and also to clarify whether development was spontaneous or was a response to provoking factors. For hemorrhagic manifestations on the skin and/or mucous membranes, the localization, prevalence, a morphological description of the elements, severity, etc. should be indicated. It is very important to note when the hemorrhagic manifestations first appeared, what, in the parents’ opinion, could have caused them, and indicate their duration . Thus, often with a deficiency of vitamin K in the body, the first manifestation of VKDK is prolonged bleeding from the site of blood collection for clinical analysis or from the site where the vaccine was administered. Taking into account the fact that blood sampling for a hemogram usually precedes vaccination, bleeding detected during this process should be an absolute indication for studying the number of platelets, their indices and coagulogram. In this case, vaccination cannot be carried out until results are obtained that exclude hemostasis disorders. It should be especially noted that our data, which are consistent with the results of studies by other authors, indicate that underestimation of the early manifestations of hemorrhagic syndrome and the lack of adequate therapy at this stage lead to a significant increase in the frequency of severe complications due to the further development of intracranial hemorrhages [11 , 17–21].
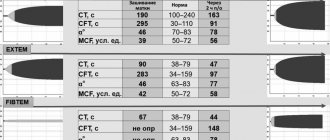
If thrombocytopenia is detected in a clinical blood test in the absence of abnormalities in the coagulogram, it is necessary to carry out differential diagnosis with a number of pathological conditions in which there is a decrease in the number of platelets (Fig. 2). If changes in the coagulogram are detected, and all laboratory indicators of primary hemostasis in a clinical blood test (platelet count, platelet indices, duration of bleeding) remain within normal limits, then we can conclude that coagulopathy occurs (Fig. 3). In this case, it is very important to differentiate at what stage of the cascade hemostasis system there is a failure. To do this, it is advisable to pay attention to the fact that in hemophilia there is a prolongation of the activated partial time (APTT) with normal indicators of the “external pathway” (prothrombin index (PTI), prothrombin time (PT), international normalized ratio (INR)) and the final stage of coagulation (thrombin time (TT), fibrinogen). Selective deficiency of coagulation factor VII is characterized by changes in the indicators of the “external pathway” (decrease in PTI, prolongation of PT and increase in INR) with normal values of the indicators of the “internal pathway” (APTT) and the final stage of coagulation (TT, fibrinogen). For a- or hypofibrinogenemia, typical changes in the coagulogram are prolongation of aPTT and TT, a decrease in PTI, an increase in PT and INR, and a decrease in fibrinogen levels [22].
In those cases when, in the absence of changes in the platelet link, disturbances are detected in the “internal pathway” (prolongation of APTT or complete absence of coagulation) and the “external pathway” (decrease in PTI, increase in PT, increase in INR or complete absence of coagulation), while As end-stage coagulation parameters (TB, fibrinogen) remain within normal limits, VCDC should be assumed (Fig. 4). These features of the coagulogram in VKDK are due to the fact that vitamin K-dependent blood coagulation factors are presented at different stages of the “internal pathway” and “external pathway” of coagulation, but do not take part in the final stage. Thus, coagulation factor IX is an obligatory component of the “intrinsic pathway”, so its deficiency will lead to a prolongation of the APTT or a complete absence of coagulation in case of severe deficiency. In turn, with insufficiency of coagulation factor VII, which is the key initiator of activation of the “external pathway,” there is a decrease in PTI, an increase in PT, an increase in INR, or a complete absence of coagulation when performing these tests, if there is a deep deficiency of factor VII. Coagulation factors II and X take part in both coagulation pathways; their deficiency will also be accompanied by changes in indicators characterizing both the “internal pathway” (prolongation of aPTT or complete absence of coagulation with deep deficiency) and the “external pathway” (decrease in PTI, increase in PT , increased INR or complete absence of coagulation). At the same time, in all these cases, the indicators of the final stage of coagulation (TV, fibrinogen) remain within normal limits, since vitamin K-dependent coagulation factors do not take part in coagulation at this level (Fig. 4).
Coagulopathy and pregnancy
As a rule, this pathology in women during pregnancy is associated with a disruption of the homeostasis system. It most often occurs against the background of the following pathological processes:
- chronic thrombophlebitis or varicose veins, which develop both during pregnancy and before its onset;
- pathological conditions of metabolic processes, such as phospholipids;
- vitamin deficiencies;
- pathologies of the circulatory system and blood formation.
Experts note a high risk of developing symptoms of coagulopathies in pregnant women who have a history of massive blood loss with complications in the form of thromboembolism in the past. Diabetics are also at risk. These patients must be under strict medical supervision of both a gynecologist and a surgeon throughout the entire period of bearing a child. They will adjust the stages of a comprehensive examination and drug therapy. As a rule, pregnant women in this risk group are sent to hospital treatment throughout the entire period before birth.
Treatment of vitamin K deficiency conditions
Timely identification of the cause of hemorrhagic syndrome makes it possible to prescribe adequate etiopathogenetic therapy in the early stages of the disease, which helps not only to stop the pathological process, but also to prevent the development of complications. In cases where we are talking about VKDK in children in the first months of life, it should be remembered that in 50–75% of cases VKDK leads to intracranial hemorrhages, accompanied by a high incidence of adverse outcomes, and in surviving children - serious complications [1–5 , 7–11, 13–21]. At the same time, most authors emphasize that intracranial hemorrhages due to vitamin K deficiency develop some time after the appearance of skin and/or mucous hemorrhages, and in some cases - against the background of ongoing bleeding from the site of blood sampling or injection, which was not noticed in a timely manner or remained underrated. In this regard, it is advisable to once again emphasize the need for an emergency search for causes even with a slight severity of hemorrhagic manifestations. In cases where, based on the analysis of clinical and anamnestic data and the results of laboratory examination, VKDK is verified, it is necessary to immediately begin replacement therapy. The drugs of choice in this case are the so-called prothrombin complexes - drugs that contain all vitamin K-dependent coagulation factors (II, VII, IX, X), as well as proteins C and S. After relief of the hemorrhagic syndrome, the planned administration of the vitamin is justified K. In cases where it is not possible to use prothrombin complex preparations, single-group fresh frozen plasma (10 ml/kg body weight) and vitamin K are administered. It should be remembered that if the effect of replacement therapy with prothrombin complex preparations or fresh frozen plasma occurs already in period of their administration, then the only synthetic analogue of vitamin K registered in our country (menadione sodium bisulfite) will manifest its hemostatic effect only after 18–24 hours. In this regard, with the development of hemorrhagic syndrome caused by VKDK, one cannot limit oneself only to the administration of menadione sodium bisulfite - simultaneous use of drugs containing vitamin K-dependent factors (or prothrombin complex, or fresh frozen plasma) is required.
How is diagnostics carried out?
Diagnosis of this pathology begins with a primary diagnosis. It is based on characteristic clinical manifestations, which can be determined by the following symptoms:
- skin and submucosal hemorrhages of varying intensity;
- hemorrhagic discharge from the intestines and bladder;
- increasing symptoms of anemia, which seriously affects the patient’s general health;
- clinical manifestations of ischemic changes in almost all internal organs;
- in the most severe cases, a picture of severe hemorrhagic shock appears.
Based on the symptoms described above, a preliminary diagnosis is made: “Coagulopathy.” After this, the attending physician prescribes medications to stabilize the serious health condition. Subsequent adjustments are made to treatment after confirmation of the diagnosis.
For this purpose, a whole range of laboratory studies is carried out. Typically, it includes the following steps:
- The total number of platelets in the blood is determined. For this pathology it should be low.
- The bleeding time is monitored; in our case, it significantly exceeds the threshold values.
- Analysis for fibrinogen content, which in this case will be reduced.
- If possible, determine the main blood factors.
- Once the diagnosis is confirmed, therapy is adjusted.
What laboratory parameters should be followed in patients with CAC/DIC?
We recommend monitoring platelet count, PT/aPTT, D-dimer and fibrinogen. Worsening of these parameters, particularly D-dimer, indicates progressive severity of COVID-19 infection and predicts that more aggressive critical care will be required; Experimental treatments for COVID-19 infection may be considered in this situation. Improvement in these parameters, along with stable or improving clinical status, provides confidence that discontinuation of aggressive treatment may be appropriate.
Basic principles of treatment
Therapy for blood clotting disorders is complex. If this pathology is not congenital, but formed as a complication of another pathological process, then treatment of the underlying disease should not stop.
To reduce the symptoms of coagulopathic bleeding, the patient is given transfusions of canned blood and its components.
//www.youtube.com/watch?v=mdvH9C5bzqo
The complex of therapeutic prescriptions includes vitamin therapy with a predominance of vitamins C, P and K. Steroid drugs are also included in the list of prescriptions.