Sickle cell anemia - what is it?
Sickle cell anemia is rightfully considered the most complex form of blood disease that is inherited. In this case, instead of hemoglobin A, the patient’s body produces hemoglobin S.
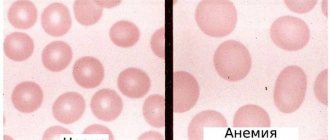
The mutated protein has an abnormal structure, which gives it characteristics that it should not normally have. Red blood cells carrying hemoglobin S change their biconcave disc shape to a sickle shape, lengthening in size. Such red blood cells are not particularly stable and are quickly destroyed in the blood vessels.
Most often, the pathology occurs in Africa, and both men and women suffer unequally. People with sickle cell anemia (both clinical manifestations of the disease and its latent course) are not susceptible to malaria.
Causes of sickle cell anemia
Sickle cell anemia is caused by a mutation in genes that is inherited. In this case, the HBV gene, which is responsible for the production of hemoglobin, is affected. As a result, a protein with an incorrect position of the beta chain is formed in the body of a sick person (glutamic acid in this chain is replaced by valine).
In this case, hemoglobin continues to be produced, but its electrical properties are disrupted. If the body suffers from a lack of oxygen, the protein changes its structure - it crystallizes and stretches into long chains, that is, it is transformed into hemoglobin S (HbS). Red blood cells react to such deformation by changing their shape. They also lengthen, which leads to thinning of their walls and destruction.
Sickle cell anemia is inherited in an autosomal recessive manner. For pathology to make itself felt, the altered gene must be received from both the father and the mother. This is the so-called homozygous form, in which hemoglobin S molecules and no others will be present in the human blood.
If the mutated gene is present in only one of the parents, then inheritance of the pathology will also occur, but the form of polymorphism will be heterozygous. In this case, the child himself will not suffer from anemia, but will become an asymptomatic carrier of the pathological gene. Hemoglobin A and hemoglobin S will circulate in his blood in different proportions. If a person is healthy, then anemia does not manifest itself in any way, since hemoglobin A is enough to cover all the needs of the body. With severe dehydration or hypoxia, anemia will make itself felt. A person who is an asymptomatic carrier of the mutated HBV gene can pass it on to their children by inheritance.
The difference between a dominant type of inheritance and a recessive one
Each genetic disease is transmitted in two types. Dominant is characterized by the fact that the disease is transmitted to a representative of each generation, regardless of gender.
Even if one parent carries the gene, 25 percent of the offspring will suffer from this pathology.
The recessive type of inheritance is characterized by the fact that the gene mutation occurs in only half of the descendants of one carrier. If one parent carries the gene for the disease, symptoms may appear within one generation.
Genetics says that recessive inheritance is more common in men. Girls can inherit it from their father. It is possible to have a son with a recessive gene from healthy parents.
The mechanism of development of sickle cell anemia
In sickle cell anemia, red blood cells stop performing their functions. Their walls become thin and fragile. In addition, the red blood cells that carry hemoglobin S are unable to transport the required amount of oxygen. They themselves cannot change shape and penetrate the smallest capillaries.
All these pathological changes in the structure of red blood cells and hemoglobin lead to the development of the following disorders:
- The lifespan of red blood cells is reduced. They die quickly and en masse in the spleen.
- Modified red blood cells precipitate and accumulate in the capillaries, blocking their lumens.
- A deficiency of red blood cells leads to their increased formation in the kidneys, and the erythrocyte germ of the bone marrow itself is degenerated.
Possible complications
The lack of timely diagnosis and qualified treatment can cause the development of the following complications and negative consequences for the patient’s health:
- liver and kidney failure;
- heart rhythm disturbance;
- myocarditis;
- pulmonary artery thrombosis;
- cerebral stroke;
- necrosis of tissues of internal organs;
- inflammation of the connective and bone tissue of the joints with their further deformation;
- pulmonary and heart failure;
- blood poisoning;
- death as a result of thrombosis of the great vessels, or internal hemorrhage.
Sickle cell anemia is a hereditary genetic disease that is dangerous due to a large number of negative consequences and complications. The disease should be under constant supervision of a hematologist. Anemia of 1st and 2nd severity reduces the patient’s quality of life, but does not lead to irreversible changes in the body.
The presence of a disease of grade 3 and 4 is the basis for an unfavorable prognosis for recovery and death at an early young age.
Article design: Vladimir the Great
How does sickle cell anemia manifest?
In a person, the symptoms of sickle cell anemia will be more pronounced the worse the health. Also, the clinical manifestations of anemia are affected by the patient’s age, living conditions and the presence of concomitant diseases. It is customary to distinguish several groups of symptoms, including:
- Symptoms of anemia caused by accelerated destruction of red blood cells.
- Symptoms of anemia caused by blocked lumens of blood vessels.
- Symptoms of anemia caused by hemolytic crisis.
The disease does not make itself felt in children under 3 months of age. Sometimes it first manifests itself at the age of 6 months.
Symptoms may include:
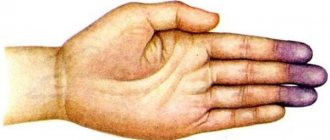
- Swelling in the area of the hands and feet, their pain.
- Muscular weakness.
- Changing the shape of the limbs.
- Retardation in physical development in terms of the formation of motor skills.
- Dry skin, paleness.
- With massive destruction of red blood cells, the skin becomes yellow, which occurs due to the release of bilirubin from the destroyed red blood cells into the blood.
Until the child reaches the age of 5-6 years, any infections can have a severe course, which is associated with pathological changes in the spleen. Its lumens become clogged with destroyed red blood cells, and the organ is unable to function normally. It is the spleen in the human body that is designed to cleanse the blood of infectious agents; lymphocytes are also formed in it. Therefore, almost any infection can lead to the development of sepsis. In this regard, parents should be especially vigilant and seek medical help for any illness.
The older the child gets, the more intense his symptoms due to chronic oxygen deprivation become, including:
- The child gets tired quickly.
- He gets dizzy regularly.
- Shortness of breath appears.
- Physical, mental and sexual development is delayed.
Women diagnosed with sickle cell disease can have children, but pregnancy comes with a number of complications.
Symptoms of sickle cell anemia in adolescents and adults:
- From time to time, painful sensations arise in different organs.
- Ulcers often form on the skin.
- Vision deteriorates.
- Kidney failure may develop.
- The structure of the bones changes.
- The joints begin to ache and swell greatly.
- Sensitivity of the limbs deteriorates and paresis occurs.
Hemolytic crisis is a dangerous condition for humans. With sickle cell anemia, it can be triggered by dehydration, severe hypothermia, excessive exercise, and mountain sickness.
The following symptoms indicate a developing crisis:
- Reduced hemoglobin levels to critical levels.
- Fainting conditions.
- High body temperature.
- Urine becomes dark in color.
Clinical picture
The clinical picture depends on a number of factors: heterozygous or homozygous state, associated infection, hypoxia, stress, pregnancy, the presence of exacerbations (crises), complications, etc.
In adults, the homozygous form, with a quiet course of the disease, is characterized by moderate anemia, which only slightly reduces the ability of patients to work. The anemia is usually normochromic, with a hematocrit value of approx. 25%; the total hemoglobin content is 80 r/l, the number of reticulocytes is 10%, irreversibly sickled erythrocytes are 12%, the average life expectancy of erythrocytes is approx. 17 days, Worsening anemia may be associated with a hypoplastic crisis (inhibition of the erythropoietic function of the bone marrow), hyperhemolytic crisis (increased hemolysis under the influence of an associated infection, hypoxia, stress factor, etc.) and sequestration crisis (decay of a significant part of red blood cells in the internal organs, in particularly in the spleen). The most specific are pain crises, characterized by acute pain in the area of erythrostasis or infarction, leukocytosis and fever. These crises are provoked by infections (in children), hypoxia, dehydration, cold, etc. Sometimes their cause cannot be determined. During pain crises, three phases are distinguished: ischemic (lasting 2-6 hours), infarction (lasting from 24 to 72 hours), microembolic (lasting up to 1 week or more). The ischemic phase is characterized by pain in the tubular bones, spine and joints.
It is reversible, especially with vigorous treatment. The infarction phase is characterized by pain in many bones and joints, fever, sweating, leukocytosis and sometimes increased anemia. During the microembolic phase, blockage of blood vessels is observed in various organs and tissues, resulting from microembolization from infused bone marrow. Depending on the location of thrombosis in S. a. Several syndromes can be distinguished - thoracic, musculoskeletal, abdominal (including hepatic), cerebral (more often in children), etc. For example, when the pulmonary capillaries are blocked, pain in the chest area, pleural effusion, and breathing are impaired.
The homozygous form in childhood is characterized by a more severe course and high mortality as a result of associated infection.
Complications of the homozygous form of S. a. are calculous cholecystitis, ulcers of the extremities (usually ankles), various bone lesions, for example, fractures, osteomyelitis, aseptic necrosis of the heads of the femur and humerus is often observed, as well as retinopathy (see), hematuria (renal origin), hyperuricemia (see Uricemia ) etc. Children may have thrombosis of cerebral vessels (see Stroke), and adults may have subarachnoid hemorrhages (see Intrathecal hemorrhages).
Wedge, picture with a combination of the heterozygous form of S. a. with beta thalassemia has the following features: splenomegaly is noted, target-shaped red blood cells appear, the content of hemoglobin F or A2 increases; Complications such as thrombosis are relatively rare.
Vascular neoplasms, vitreous hemorrhages, ischemia and retinal detachment are more common in patients with SC hemoglobinosis.
How to detect sickle cell anemia?
Diagnosis of sickle cell anemia begins with questioning the patient’s complaints and collecting an anamnesis. The symptoms that the disease gives are characteristic of many pathologies, so laboratory tests are required.
The main diagnostic methods include:
- Blood sampling for clinical analysis. In this case, a decrease in the level of red blood cells and hemoglobin in the blood will be detected to levels less than 3.5-4.0 * 1012/l and 120 g/l, respectively.
- A biochemical blood test allows you to diagnose increased levels of bilirubin and free iron in the blood.
Additional diagnostic procedures include:
- Performing a blood test using sodium metabisulfite. When interacting with this substance, red blood cells release oxygen, after which their sickle shape can be visualized.
- Treatment of blood with buffer compounds. HbS is poorly soluble in them.
- Performing hemoglobin electrophoresis, which makes it possible to visualize modified red blood cells. This method also allows you to distinguish a heterozygous mutation from a homozygous one.
Other examination techniques that may be required:
- Ultrasound examination of the liver and spleen, as well as other internal organs to detect pathological changes in them.
- Performing x-rays of the bones of the skeleton and spine.
Diagnostics
Sickle cell anemia is a dangerous pathology of the blood and hematopoietic system, which requires a comprehensive diagnosis of the whole organism.
A patient who has signs of this disease must undergo the following types of examination:
- initial examination by a doctor who listens to current complaints and symptoms, palpates the abdominal cavity, chest bones and limbs, and makes a referral for instrumental and laboratory diagnostics;
- donation of venous blood for its biochemical analysis;
- collection of capillary blood for clinical research and isolation of red blood cells;
- providing morning urine to determine the patient’s general health;
- ECG to ensure the stability of the heart muscle;
- blood pressure measurement;
- MRI of the whole body to evaluate the health of blood vessels, as well as tissue in internal organs that may have been damaged by blood clots and increased fragility of red blood cells.
Based on the results of a diagnostic examination, the attending physician confirms the presence of sickle cell anemia, or refutes the pathological condition of the blood and hematopoietic system. In public hospitals, examinations are carried out free of charge. In a medical institution with private ownership, the average cost of this diagnosis will be from 4,000 to 6,000 rubles.
Treatment of sickle cell anemia
Treatment of sickle cell anemia involves influencing the symptoms of the disease and is also intended to prevent the development of complications.
Therefore, when conducting therapy, you need to focus on the following principles:
- Replenish the lack of red blood cells and hemoglobin.
- Relieve painful sensations.
- Eliminate excess iron from the body.
- Eliminate the consequences of a hemolytic crisis.
To normalize the level of red blood cells and hemoglobin, a transfusion of red blood cells may be required. Alternatively, the patient is administered cytostatics (hydroxyurea), which increase the level of hemoglobin in the blood.
To relieve pain in the patient, he is prescribed analgesic drugs (Morphine, Tramadol, Promedol). When the disease is acute, it is necessary to administer drugs intravenously. After the exacerbation is relieved, they are administered orally.
You can remove excess iron from the body through the use of certain medications, for example, using Deferoxamine.
During a hemolytic crisis, the patient is prescribed oxygen therapy, adequate replenishment of fluid reserves, analgesics, drugs to relieve seizures, etc.
When an infection enters the body, the patient is prescribed antibiotics, which help prevent sepsis. These may be drugs such as Cefuroxime, Amoxicillin, Erythromycin.
All patients with sickle cell anemia must adhere to the following medical recommendations:
- Lead a healthy lifestyle and completely give up bad habits.
- Avoid climbing to heights.
- Avoid excessive physical activity.
- Drink enough water.
- Avoid overheating and hypothermia.
- Eat healthy foods.
Prevention measures
The prognosis for patients with sickle cell disease is not always good. If children get the homozygous form of the disease, they die from infection or from blocked blood vessels.
For carriers of a defective gene prognosis are more successful, but they must comply with several principles, which include:
- Choosing a place of residence in which a temperate climate prevails, and the altitude above sea level does not exceed 1.5 thousand MASL;
- avoid alcohol and drugs;
- avoid smoking;
- Choosing a profession that does not involve high stress, contact with toxic substances and operating rooms with high air temperatures;
- Drink plenty of liquids a day, at least half a liter.
Both parents are considered before the baby is born. An inherited disorder may be discovered if a mutant of sickle cell disease is discovered after studying the genetic material.
Detection of mutagen in the early stages of embryo development occurs using modern methods.
A positive test result is a problem for expectant parents. Ultimately, they may appreciate the importance of deciding to terminate the pregnancy in a timely manner or hope for the birth of a healthy child who carries the gene without symptoms of anemia.