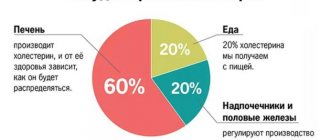
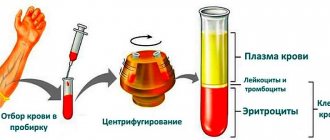
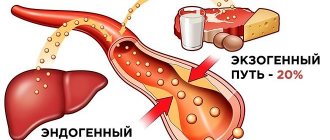
This organic fat-like substance performs many important functions. Without it, the synthesis of hormones, the absorption of most vitamins, the formation of bile acids, and hematopoiesis are impossible. Most of the cholesterol is produced inside the body, about 20% comes from the outside, with food. An increase in the level of this compound is fraught with the development of atherosclerosis and other diseases. Excess cholesterol fills the lumen of blood vessels and settles in the gallbladder in the form of stones.
Complexes with this research
Expanded hospital complex Expanded infectious screening for prevention and hospitalization RUB 7,700 Composition
Blood biochemistry. 13 indicators Optimal biochemical blood test RUR 3,490 Composition
Male check-up No. 1 39 studies for annual preventive examination RUR 18,570 Composition
IN OTHER COMPLEXES
- Biochemistry of blood. 8 indicators 990 R
- Pregnancy planning. Clinical indicators 6,630 R
- Preventive check-up RUB 11,960
- Examination during pregnancy. 3rd trimester 9,620 RUR
- Cola and chips RUB 3,020
Treatment and prevention
Today, high cholesterol in children is not uncommon. If this pathological condition is identified during an examination, the fight against it must begin as early as possible. Normalizing high cholesterol in children is somewhat easier than in adults. Before the onset of puberty, hypercholesterolemia can be easily corrected by normalizing the diet physical activity (race walking, running, swimming, cycling, skiing) to the daily routine
Forming healthy eating habits is an important task for parents.
Sometimes, if the above recommendations are followed, serum cholesterol levels do not decrease. Then the need for drug treatment arises and the question arises: “Can I take statins?” The instructions for drugs in this group state that they are contraindicated for children under 18 years of age due to the fact that there is no data on their safety and effectiveness in this age group.
Pediatricians recommend medicinal treatment of high cholesterol in children using the drug “Policonazole”. It is made from natural raw materials - sugar cane stalks. The medicine helps eliminate the imbalance between LDL and HDL and reduce total cholesterol levels. To achieve the desired result, taking Polyconazole must be combined with proper nutrition and physical activity.
Prevention of hypercholesterolemia involves maintaining a healthy lifestyle. This requires a diet and nutrition that contains a minimum of harmful fats. It is also necessary to monitor children’s body weight and teach them to systematically play sports. It should be remembered that the foundation for most diseases that occur in adulthood is laid in childhood!
When should you take a total cholesterol test?
- Diabetes;
- Obesity;
- Thyroid diseases;
- Myocardial infarction or stroke in close relatives;
- Long-term smoking;
- Alcohol abuse;
- Chronic liver and kidney diseases;
- Poor diet with excess fatty foods;
- The presence of a chronic inflammatory process;
- Comprehensive assessment of health indicators in people without risk factors for cardiovascular diseases;
- Assessment of changes in cholesterol levels over time due to diet and/or treatment.
Cholesterol and baby
The child’s body is actively developing, new cells and hormones are formed, and the nervous system is strengthened. For all this you need a sufficient level of cholesterol.
A child's metabolism is always very active and fast, as this is normal for a growing body. The logical conclusion follows from this: a healthy child should have high cholesterol levels.
Important:
Low cholesterol levels in children are a reason to consult a doctor and determine whether there are metabolic disorders.
Elevated cholesterol levels in children are completely normal. Only with a high level of cholesterol will a child develop good nervous tissue, hormones, and cellular immunity. A high level indicates that the body is developing, and this is not a reason for panic, but quite the opposite.
High levels are not a disease. But when it is low, then there is a risk of developing many diseases, developmental delays, and metabolic disorders. This indicator should be a reason to consult a doctor.
Detailed description of the study
Cholesterol is a fat-soluble compound that performs many functions necessary for normal cell function. It serves as an important component of the cell membrane: it helps maintain the structure of the membrane and also changes its viscosity.
The functions of cholesterol in the body are diverse. It is a precursor molecule in the synthesis of vitamin D, steroid hormones (eg, cortisol, aldosterone, and adrenal androgens), and sex hormones (eg, testosterone, estrogens, and progesterone). Cholesterol is also part of bile salts, which are needed to digest food and improve the absorption of fat-soluble vitamins A, D, E and K.
Maintaining cholesterol levels in the blood occurs largely due to its synthesis in liver cells (70-80%). Therefore, the content of this substance in the body depends on the condition of the liver, and is also predetermined by genetic factors. Cholesterol levels are not very different between men and women, but may increase with age.
In the liver, cholesterol combines with carrier molecules (lipoproteins) and enters the blood, then into cells and tissues to perform its functions. Although this compound plays an important role in the body, it can also have harmful effects when present in excess in the blood. This condition is called hypercholesterolemia.
Hypercholesterolemia increases the risk of cardiovascular disease and its complications, such as myocardial infarction or stroke. A predisposing disease for this is vascular atherosclerosis, that is, the formation of cholesterol-containing plaques that can block the lumen of the arteries and increase the risk of blood clots. The presence of high sugar (glucose) in the blood, impaired liver function and decreased production of thyroid hormones contribute to increased cholesterol in the blood and the development of atherosclerosis.
Hypercholesterolemia usually does not show any symptoms until the atherosclerotic plaque causes complications in the heart (myocardial infarction) or blood vessels: stroke, dizziness due to impaired blood flow in the vessels of the neck, intermittent claudication due to damage to the arteries of the lower extremities. Some people may notice accumulations of fatty tissue under the skin (xanthomas) and yellowish plaques on the eyelids (xanthelasmas). These benign formations are usually considered a cosmetic defect, but they reflect a disorder in the metabolism of cholesterol in the body.
Studying the level of cholesterol in the blood is important for the timely detection of fat disorders in the body and the prescription of treatment to prevent damage to the blood vessels of the heart and other various organs.
Causes of Low Cholesterol
Dr. Shishonin provides many arguments in favor of the fact that elevated cholesterol levels in children are a normal, healthy phenomenon. But what to do if the analysis reveals a reduced level of this indicator? This is a very alarming symptom, and it manifests itself for several reasons:
- Genetic predisposition. As you know, heredity can play a role in many characteristics of the body. There are cases when, due to genetic predisposition, low cholesterol levels in a child are normal, but in this case you should still see a doctor.
- Poor nutrition. For normal growth and development, a child needs a balanced diet. You can’t limit him in proteins, fats, carbohydrates and calories, but you should keep an eye on exactly what foods he eats. Include more dairy products rich in highly digestible fats in your child’s diet and limit fast food and other “junk” foods.
- Liver diseases. As you know, the liver is the main organ that produces cholesterol from fats. If the liver is not in order, it means that a sufficient amount of cholesterol, so necessary for the normal development of the child, does not enter the blood. The consequence is metabolic disorders and further complications.
- Thyroid diseases. It is necessary to constantly keep under control those organs that are responsible for the production of hormones, since hormonal disorders are difficult to reversible. Provide sufficient amounts of iodine-rich foods in your child's diet and do not forget to undergo regular examinations with an endocrinologist.
Important:
A low cholesterol level always indicates the presence of some kind of developmental disorder.
At the same time, the following are considered normal indicators for children:
- Newborn – 53–135 mg/l (1.37–3.5 mmol/l).
- Up to 1 year – 70–175 mg/l (1.81–4.53 mmol/l).
- From 1 year to 12 years – 120–200 mg/l (3.11–5.18 mmol/l).
- 13–17 years – 120–210 mg/l (3.11–5.44 mmol/l).
Thus, elevated cholesterol levels are normal for a child. The alarm should be raised only if this indicator decreases and goes beyond the normal range. This indicates the presence of metabolic disorders and cholesterol metabolism.
Hereditary hypercholesterolemia in children
Hereditary hypercholesterolemia in children
Zubov L.A., Lebedev A.V., Tril V.E., Northern State Medical University, Arkhangelsk
Lipid metabolism disorders can be grouped according to the predominant biochemical phenotype: hypercholesterolemia, combined increases in cholesterol and triglycerides, hypertriglyceridemia, hypolipidemia, secondary dyslipidemia.
Familial combined hyperlipidemia Hypertriglyceridemia associated with hypercholesterolemia is most often associated with a combined increase in low-density lipoprotein (LDL) and very low-density lipoprotein (VLDL). Prevalence in the population is 1:50 – 1:300. The incidence in patients with coronary heart disease (CHD) is 15-30%. Various types of hyperlipidemia occur within the same family. Often accompanied by a decrease in high-density lipoprotein cholesterol (HDL-C). The type of inheritance is autosomal dominant. Familial hypertriglyceridemia The concept combines several types of genetic disorders. The most common disease is associated with excessive synthesis of hepatic triglycerides (TG). The type of inheritance is autosomal dominant, occurs in 15% of patients with premature ischemic heart disease, and is often accompanied by components of the metabolic syndrome (obesity, impaired glucose tolerance, hyperuricemia). A more severe form of familial hypertriglyceridemia, associated with deficiency of lipoprotein lipase and apolipoprotein C II, has an autosomal recessive mode of inheritance and is quite rare (less than 1: 10,000). The TG level rises to 10.0 mmol/l, and the risk of recurrent pancreatitis is increased. Hereditary hypercholesterolemia (HHC) Hereditary hypercholesterolemia is an autosomal dominant, monogenic disease, the development of which is caused by a violation of the structure or function of receptors for low-density lipoproteins (LDL) on somatic cells (liver and others) and/or their number [5,13] ( the classic form of NHHS [5]), or disturbances in the structure of the molecule of apolipoproteins B-100 and C (apo B-100 and C, the protein components of lipoproteins). Receptor disruption is determined by the presence of a mutated gene in the LDL locus on the short arm of the 19th autosomal chromosome or “apo B-3500 mutation” - a violation of the structure of apo B-100 at position 3500: Arg3500→Gln [17] (chromosome 2); apo C mutations (chromosome 19) [3,4,12,14-16,18] with an autosomal dominant mode of inheritance. The presence of mutations in the LDL receptor locus occurs in 0.1% of individuals in the general population and 1% of patients with premature coronary heart disease (CHD) [11,14]. Moreover, the prevalence in the population of heterozygous forms, when a child receives mutant genes from one of the parents, (1:500) is much higher than homozygous forms (1:106) [3]. Homozygous familial hypercholesterolemia is rare. The probability of two unrelated heterozygotes marrying is 1 in 250,000, and the chance of them having a homozygous child is one in four, based on this, its theoretical frequency is also one chance in a million. The likelihood of marriage between heterozygotes increases significantly with consanguineous marriages. About 700 types of these mutations are known. Apo B and apo E mutations are observed less frequently. The average incidence of familial hypercholesterolemia is 1:500 [19]. Understanding the metabolic basis of hypercholesterolemia relies on knowledge of the type of genetic mutation of the LDL receptor. Depending on the nature of the genetic defect, the disease is classified as receptor-negative, receptor-defective, or receptor-internalizing forms. [7]. The main disorders are impaired catabolism of LDL by the liver and other tissues through LDL receptors that bind to apo B. With pathology of LDL receptors, this causes an increase in the level of LDL in the blood and an increase in the circulation time of this type of lipoprotein, which ultimately leads to the formation of altered, modified forms of LDL, which are highly atherogenic [3]. Homozygous forms of NHC differ from heterozygous ones in the complete absence of LDL receptors on the surface of cells and removal of LDL from the bloodstream in this way, and longer circulation of their peripheral bloodstream as a result of these changes (capture of LDL by cells occurs in a non-receptor manner) [5], whereas in heterozygous forms, the number of receptors is reduced by about half. In the presence of the apo B-3500 mutation, the apo B-100 protein does not bind to the receptor. This mutation occurs at a frequency of approximately 1 in 600 in the general population, although it does not usually cause particularly severe hyperlipidemia. Almost 4% of individuals with clinical familial hypercholesterolemia may have a defect in apolipoprotein B. This leads to the development of similar disorders and increased levels of total cholesterol (TC) and LDL cholesterol in the blood, ultimately contributing to the activation of atherosclerotic changes in the cardiovascular system. Histological manifestations of this process in the aorta and large arteries will be: lipid saturation of the vessel wall, vasa vasorum lipoidosis, an abundance of foam cells and cholesterol crystals in the intima and media. With further progression, these disorders lead to the formation and growth of atherosclerotic plaques. Atherosclerosis develops when cholesterol levels in the blood exceed the body's ability to remove cholesterol from the bloodstream, thereby leading to fat deposits in the vessel wall. Clinical manifestations correspond to the level and prevalence of pathological disorders. The most striking example of the early occurrence of atherosclerosis is its development in children with homozygous NHHS. Clinical observation Girl V., 14 years old, has been observed for 6 years for hereditary hypercholesterolemia. Plaque formations in the area of the left patella, oval in shape, beige in color, 0.3-0.6 cm, first appeared at the age of 8. A biochemical study revealed hypercholesterolemia: total cholesterol 12.3 mmol/l, 11.4 mmol/l, 14.0 mmol/l; β - lipoproteins 111 IU, 110 IU (N up to 55 IU). After 1 year, tendon xanthomas appeared in the area of the tendons of the metacarpophalangeal joints on the left and Achilles on both sides. TC – 10.1 mmol/l, β-lipoproteins 130 IU. During examination in 2000: total cholesterol 12.0 mmol/l; β - lipoproteins 87.3 IU, 70 IU; a-lipoproteins 0.6 mmol/l; TG 1.31 mmol/l; atherogenic index 16.9 units. Family history: maternal grandfather – xanthomas, ischemic heart disease, high cholesterol (died), grandmother is healthy; the mother had xanthomas, high cholesterol, varicose veins, obesity, a brain tumor (died at the age of 30), the mother had 3 brothers and 1 sister, all healthy; father – coronary artery disease, high cholesterol, chronic alcoholism, cirrhosis of the liver (died); no lipid metabolism disorders were observed in relatives on the father's side; The girl's older brother is healthy. According to the family history, it can be concluded that the mother and maternal grandfather had hereditary heterozygous hypercholesterolemia; the increase in blood cholesterol levels in the father is most likely of a secondary or polygenic nature. After establishing a clinical diagnosis, therapy with a drug from the group of HMG-CoA reductase inhibitors (pravastatin) was prescribed at a dose of 5 mg per day continuously. Periodically courses of lipotropic and hepatotropic drugs. Regular blood lipid monitoring and examination are carried out in a children's regional hospital. Statin therapy is well tolerated and allows maintaining blood cholesterol levels at 8.0 – 9.1 mmol/l. Clinical and laboratory changes differ in severity in patients with homozygous and heterozygous forms. Skin and tendon manifestations On the skin of patients with NHHS, pathognomatic signs of lipid metabolism disorders are observed: xanthomas and xanthelasmas (deposition of cholesterol esters in the thickness of the skin). Xanthomas (xanthomatous plaques) are divided into several types: tendinous, eruptive, tuberous (5). Tendon xanthomas - thickening in the area of the tendons (Achilles, tendons of the metacarpophalangeal joints, extensors of the palms and feet - the first two types of localization are most common), are better visible when flexed, sometimes accompanied by signs of inflammation. The skin covering the xanthomas is of normal color. Cholesterol deposition occurs deep in the tendons and most of the swelling represents fibrosis, as a result of which xanthomas feel hard when palpated. Achilles tendon xanthomas can be clearly visible due to the significant size of the thickening and swelling and resemble tubercles. But they can be small, then they are determined only by palpation of the tendon. Xanthomas on the dorsum of the hand also usually resemble bumps or nodules, and since they often overlap the joints of the hand (especially when clenching the hand into a fist), they can resemble bone in density - doctors can often miss them. Hands should be examined with fingers spread apart. For diagnosis, the ability of xanthoma to move backward from the joints and from side to side is used. Eruptive xanthomas are small xanthomas (usually no larger than a millet grain), usually located in groups on the skin of the torso, knees, elbows, buttocks, palms and even on the mucous membrane of the oral cavity. Tuberous xanthomas are tuberous in shape, preferably located on the elbows. Xanthelasmas are yellowish nodules located under the lower and upper eyelids of the eyes. Eye symptoms Lipemic arc along the periphery of the cornea (Corneal arcus) Tendon xanthomas are one of the most important diagnostic signs, as they are rarely found in other diseases. These are localized infiltrates of lipid-containing foam cells that histologically resemble atheroma. Lipemic arches and xanthelasmas are not as specific for familial hypercholesterolemia, but their appearance in this disease is usually observed earlier in life than in the more common polygenic type of hypercholesterolemia. And in many patients with a heterozygous form of hereditary hypercholesterolemia with pronounced tendon xanthomas, lipemic arches are noted only in the late stages of the disease and xanthelasmas never develop. Therefore, all patients with hypercholesterolemia should be screened for the presence of tendon xanthomas, regardless of the presence of arcus corneale or xanthelasma. Since this examination method can be a kind of screening for NHHS. Homozygous forms of the disease are characterized by a combination and early appearance of xanthelasmas and various types of xanthomas. Flat orange-yellow xanthomatous plaques appear on the buttocks, in the interdigital area, on the palms and the front of the knees. In heterozygous NHHS, isolated tendon xanthomas are more common after the age of ten. Hyperlipidemia (HLP) There is an increase in the level of total cholesterol, LDL cholesterol, and less often TG. According to the Fredrickson D. and WHO classification, hyperlipidemia observed in children with NHHS will correspond to classes II a (more often observed) and II b classes (Table 1). Both types of HLP have a pronounced atherogenic orientation. Table 1. Characteristics of hyperlipidemias occurring in NHHS.
| GLP type | LDL cholesterol | TG | VLDL cholesterol | Clinic | |
| II a | ↑↑ | ↑↑ | Norm | Norm | Xanthomas |
| II b | ↑↑ | ↑↑ | ↑↑ | ↑↑ | Xanthomas, xanthelasmas |
The concentration of blood cholesterol in heterozygous familial hypercholesterolemia increases from childhood. If the TC level is more than 7.8 mmol/l [3.8] in childhood, it is necessary to exclude NHHS, and according to foreign authors, during observation and additional examination of children with TC levels of more than 6.7 mmol/l, in a large percentage of cases it is possible to identify hereditary disorders lipid metabolism [10]. Hypercholesterolemia in children can be observed from the moment of birth and throughout life; total cholesterol levels usually increase from 8.0 to 12.0 mmol/l. An increased concentration of total cholesterol can be detected even when examining the blood of a newborn (in a patient with NHHS it will significantly exceed the normal concentration of cholesterol in the blood serum of the umbilical cord 1.7-2.0 mmol/l However, screening for total blood cholesterol at this age is not recommended , since LDL, which is the dominant lipoprotein in the blood of the fetus, can cause an increase in cholesterol concentrations much more often than familial hypercholesterolemia.Serum cholesterol in healthy children in the UK and the USA, according to foreign authors [10], increases in the first year of life to the average values of 4.1 mmol/l (95th percentile – 5.2 mmol/l) and remains at this level until the beginning of the second decade of life. Moreover, the indicators before puberty in girls and boys do not differ significantly. This is what justifies the decrease in the diagnostic threshold for childhood NHHS for total serum cholesterol above 6.7 mmol/L. Upon further examination of children from this group, the diagnosis of NHHS is confirmed in 95%. Blood cholesterol levels in people with familial hypercholesterolemia, as in the general population, increase with age. In general, values in heterozygous familial cholestrolemia are usually twice those in the absence of an LDL receptor mutation. In adults with heterozygous NHCH, cholesterol is usually in the range of 9.0 – 14.0 mmol/l. In families of identified probands with NHHS, during a single examination of children, the detection of relatively low blood cholesterol concentrations, such as 5.2 - 5.5 mmol/l, does not at all exclude the diagnosis, especially if the family is already on a lipid-lowering diet. In this case, repeated studies of the lipid spectrum are necessary. Thus, the recommended criteria for the heterozygous form of NHHS are: serum cholesterol concentration greater than 6.7 mmol/L in children under 16 years of age or more than 7.5 mmol/L in adults, plus tendon xanthoma in the patient or first- or second-degree relative. A small number of patients also experience elevated blood triglyceride levels, although rarely above 4.0 mmol/L. The presence of elevated TG aggravates the course of the disease and worsens the prognosis. Hypertriglyceridemia is often observed in overweight patients with NHHS. The total cholesterol level often exceeds 20.0 mmol/l. Obesity is generally rare in familial hypercholesterolemia compared to almost all other hyperlipidemias. Hypertension and diabetes mellitus are also relatively uncommon in NHHS. The homozygous form of familial hypercholesterolemia is one of the most severe monogenic disorders of lipid metabolism. In children with homozygous forms of NHHS, the increase in the level of blood cholesterol is more pronounced and exceeds 15.0 – 18.0 mmol/l, in some cases it can reach 25.0 mmol/l [3] and even 40.0 mmol/l [9] . The clinical manifestation will also be massive cutaneous and tendon xanthomatosis. Xanthomas can be detected in the first year of life. Triglyceride levels are normal or even low or low. Xanthomas develop at an earlier age. In addition to pronounced tendon xanthomas, orange-yellow skin flat xanthomas appear in the buttocks, interdigital tissue, palms and knees. Polyarthralgia is often observed. Identification of each new case of familial hypercholesterolemia makes it possible to identify affected relatives. This approach, known as cascade family screening, appears to be better than general or selective screening. Early development of atherosclerosis and coronary artery disease A feature of patients with familial hypercholesterolemia is the early development of cardiovascular diseases of an atherosclerotic nature. If patients are left untreated, more than half of men and about 15% of women with the heterozygous form die before the age of 60 years. The most serious clinical manifestations of NHHS are early signs of coronary heart disease, atherosclerotic lesions of the heart and blood vessels. In heterozygous forms of NHHS, signs of IHD are detected by the age of 40–50 in men and a decade later in women, and in the case of a homozygous form of the disease, atherosclerotic changes are usually detected before the age of 20 [9], and cases of early death from IHD are observed even before 30 years [3]. In some cases, familial hypercholesterolemia appears to be particularly destructive, causing cardiovascular disease in men from the age of 20, and in women even before menopause. In other families, heterozygous men are not affected until late middle age, sometimes even into old age, and women live into old age with few symptoms of the disease. There has been much debate as to why some families with NHHS are more susceptible to CHD than others. The type of mutation is important, as some mutations are more likely to impair LDL uptake than others. HDL cholesterol, being a protective factor, also determines the likelihood of developing coronary heart disease in familial hypercholesterolemia. Serum HDL cholesterol is often below the required protective level in familial hypercholesterolemia. If this fact persists for a long time, the prognosis in a large number of cases is unfavorable. Clinical manifestations of atherosclerosis in homozygous forms are often detected in early childhood and even infancy. Supravalvular aortic stenosis can cause sudden death. Many homozygotes develop angina pectoris during physical activity in childhood due to aortic stenosis and the presence of coronary atheroma. Myocardial infarctions have been described as early as 2 years of age, and life expectancy usually does not exceed the early third decade. Homozygous familial hypercholesterolemia is always severe, but the worst prognosis appears to occur when both LDL mutations occur in a type that completely eliminates the appearance of LDL receptors on the cell surface. Treatment and observation The main goal in the treatment of NHCS is control and, if possible, normalization of blood lipid levels, as well as the exclusion and prevention of all risk factors for coronary artery disease, diseases and pathological conditions that lead to even greater destabilization of lipid metabolism and aggravation of the course of NHHS. Basic principles of treatment: 1. Diet therapy. 2. Increasing physical activity. 3. Pharmacotherapy. 4. Methods of extracorporeal treatment. 5. Genetic engineering methods 6. Surgical treatment methods Diet therapy is the first step and a necessary component in the treatment of any type of hyperlipidemic conditions, and NHHS in particular. Basic rules of diet therapy: constant use with changes in the eating habits of the whole family and limiting fat consumption to a level that is safe for a growing and developing body while meeting the need for proteins, vitamins and microelements. It is recommended to limit fat intake to 30% of total calories, but not less than 20%. Daily cholesterol intake is no more than 300 mg [20]. If ineffective, fat consumption is limited to 25% of total calories. Daily cholesterol intake is no more than 200 mg. Some recent publications express an opinion about the possibility of a more stringent reduction in cholesterol in the diet, but here it is necessary to note the importance of a balanced position in relation to limiting its intake into the child’s body. A lack of it in the diet is no less dangerous than an excess, and the exact percentage of fat from the total calorie content that provides the physiological needs of the child’s body at various stages of its development is a subject for study and discussion. Cholesterol is necessary for the processes of growth and development of the body, differentiation and formation of cell structure, and the functioning of membrane ion pumps. It is also used for the synthesis of steroid hormones (including sex hormones), metabolic processes of large amounts of vitamins, etc. Often, to calculate the diet and correct age-related needs for nutrients, vitamins and minerals, the help of an experienced nutritionist is required.
Regarding the diet of foods, you can give the recommendations of the European Society for the Study of Atherosclerosis: 1. Recommended foods. Wholemeal bread, rice, crispbread, milk and low-fat cheeses, all fresh or dried, frozen fruits and vegetables, fish, fish oil, oriental sweets, nougat, candies, peppers, mustard, spices, walnuts, almonds. 2. Products whose consumption should be limited: Pasta, fried potatoes, potato flakes, cakes, biscuits, halva, peanuts, pistachios, egg yolk. 3. Products whose consumption is not recommended. Butter pastries, cream, fatty cheeses and yoghurts, caviar, fried fish, duck meat, all types of fatty meat, sausages, ham, poultry skin, lard, palm oil, butter, hard margarine, mayonnaise, chips, ice cream, dumplings, chocolate. It is recommended to limit the content of table salt, which reduces the activity of enzymes responsible for the breakdown of fats and contributes to an increase in blood pressure. Salt also increases the permeability of the walls of blood vessels. Prescribing dietary therapy alone for NHHS usually does not allow controlling lipid profile indicators and requires medicinal intervention. Drug therapy is prescribed if diet therapy is ineffective for 6–12 months while the level of LDL cholesterol in the blood remains high (more than 4.91 mmol/l) in children over 10 years of age [20]. The drugs of choice are bile acid sequestrants (cholestyramine and colestipol), which bind and increase the excretion of bile acids from the body. According to A.N. Klimov, they are indicated only for the heterozygous form of NHHS; for the homozygous form, their use is completely ineffective. Treatment with these drugs is poorly tolerated by children and therefore they are not always prescribed, since if the child has a negative attitude and refuses to take the drugs, especially in adolescence, the drugs do not perform the function of preventing cardiovascular diseases. The second group of drugs used in the treatment of NHCS are HMG-CoA reductase inhibitors (statins). The use of this group of drugs in childhood is a debated issue, but there is already a sufficient number of studies confirming the effectiveness and safety of their use [21,22] subject to certain rules: start treatment with the lowest therapeutic dose with a gradual increase in dosage in case of insufficient control of the lipid profile and absence side effects. Monitoring of liver function tests and hemogram parameters is necessary. It should be used with caution in teenage girls if pregnancy is possible, as adverse effects on the fetus cannot be ruled out. Contraindicated: with severe liver dysfunction, renal failure, pregnancy, lactation, myopathies. Statin drugs have represented significant advances in the management of familial hypercholesterolemia in adults. Most heterozygotes for familial hypercholesterolemia may have serum cholesterol concentrations below 7.0 mmol/L, and some even below 5.0 mmol/L. Sometimes the effect of statin therapy alone is insufficient. In this case, adding a drug from the group of bile acid sequestrants is the most logical approach. The development of more potent statins or the use of the cholesterol absorption inhibitor ezetimibe may be alternatives to this approach in the future. The penetrance of familial hypercholesterolemia, assessed in the CAD risk concept, may be useful in making decisions about the age of initiation of statin treatment. Typically, in heterozygous males, statin treatment should begin in late adolescence. But if the family history is particularly unfavorable, then earlier therapy may be prescribed. In girls, the start of treatment may be postponed to a later date. The issue of birth control while taking statins should be discussed with them. In the future, when planning a pregnancy, women can stop taking statins when they are trying to get pregnant, as well as during pregnancy and lactation. Nicotinic (up to 7 mg per day) and lipoic acids, membranotropic and hepatoprotective drugs containing polyunsaturated phospholipids (Essentiale, Lipifarm, Lipostabil), antioxidants (vitamin E, beta-carotene, vitamin C), sorbents, and polyunsaturated fatty acid preparations are also used. (eikonol, gamma-linoleic acid), multivitamin preparations. Patients with homozygous HCS are usually refractory to diet therapy and traditional lipid-lowering drugs. The most potent statins can lower serum cholesterol in up to 30% of individuals with homozygous familial hypercholesterolemia. The drug ezetimibe may provide a further 20% reduction. However, even then significant hypercholesterolemia persists. The treatment of choice for such patients remains lifelong LDL apheresis or plasmapheresis in combination with HMC-Co-A reductase inhibitors (statins), liver transplantation, or gene therapy. The last two methods are practically not used due to technical complexity and high lethality, although they give a certain effect. LDL apheresis or plasmapheresis is the best treatment. Typically the procedure should be performed every 2 weeks. A new and promising direction in the treatment of NHC is genetic engineering methods. NHHS may become one of the first genetic diseases that will be radically treated with this method. Literature 1. Gratsiansky N.A. Blood cholesterol, coronary heart disease, nutrition and some other “coronary” risk factors. When and why you need to determine cholesterol in children and adolescents. Moscow - 2000 2. Janashia P.Kh., Nazarenko V.A., Nikolaenko S.A. Dyslipoproteinemia: clinical picture, diagnosis, treatment: Textbook. allowance. – M.: Ros. state honey. University, 2000. 3. Klimov A.N., Nikulcheva N.G. Metabolism of lipids and lipoproteins and its disorders. "Pi-ter", St. Petersburg. – 1999. – 512 p. 4. Kuchinsky A.I., Isakov V.A. Clinical use of the chain reaction of DNA synthesis to analyze the structural features of the apoprotein B and C III genes in patients with coronary artery disease. The role of heredity and other risk factors in the development of cardiovascular diseases. Saint Petersburg. – 1992. – P. 23 – 27. 5. Lipovetsky B.M. Clinical lipidology. Saint Petersburg. – 2000. – 119 p. 6. Lipovetsky B.M. Familial hypercholesterolemia in parents and children. Frequency of inheritance, clinical manifestations, severity of atherogenic shift. Russian cardiological journal. – 2003. – No. 2. 7. A.V.Susekov, L.A.Kotova, M.Yu.Shcherbakova, A.O.Eliseev, Z.A.Akhmedova, M.G.Tvorogova, V.V.Kukharchuk . Modern approaches to the treatment of hereditary homozygous hypercholesterolemia in children. – http: www.incart.spb.ru/vestnic. 8. Shcherbakova M.Yu. Lipid metabolism disorders. Pediatrics. – 2000. – No. 4. – P. 76-80. 9. Shcherbakova M.Yu. Dyslipoproteinemia. Attending doctor. – 1999. – No. 7. 10. Durrington P. Dislipidemia. – The Lancet. – 2003. – August 30, Vol. 362. 11. Genest JJ Jr., Martin-Munley SS, McNamara JR et al. Familial lipoprotein disorders in patients with premature coronary artery disease. Circulation – 1992. – Vol. 85 – P.2025 – 2033. 12. Goldstein J., Brown M. Familial hypercholesterolemia. In: Stanbury JB, Wyngaarden JB et al. The metabolic basis of inherited disease, 5th ed. New York. – 1983. – P.672 – 712. 13. Goldstein J., Brown M. Lipoprotein receptors, cholesterol metabolism and atherosclerosis. –Arch. Pathol., – 1975. – Vol. 99 – P. 181 – 184. 14. Goldstein J., Shrott HG, Hazzard WR, Biernan EL, Motulsky AG Hyperlipidemia in coronary heart disease. Genetic analysis of lipid levels in 176 families and delineation of a new inherited disorder, combined hyperlipidemia. J. Clin. Invest. – 1973. – Vol. 52 – P. 1544 – 1568. 15. Fredrickson DS, Levy RI, Lees RS Fat transport in lipoproteins: an integrated approach to mechanism and disorders. N.Engl. J. Med. – 1967. – Vol. 276 – P. 34 – 44, 94 – 103, 148 – 156, 215 – 225, 273 – 281. 16. Brown M., Goldstein J., The LDL receptor concept: clinical and therapeutic implications. In: Stokes J. III, Mancini M. ed. Hypercholesterolemia: clinical and therapeutic implications. Atherosclerosis Reviews. – 1987. – Vol. 18 – P. 85 – 94. 17. Inazu A., Brown M., Hesler CB Increased high-density lipoprotein levels caused by a common cho-lesteryl-ester transfer protein gene mutation. – N. Engl. J. Med. – 1990. – Vol. 323 – P. 1334 – 1338. 18. Innerarity TL, Weisgraber KH, Amold KS, et al. Familial defective apolipoprotein B-100: low density lipoproteins with abnormal receptor binding. Proc. Natl. Acad. Sci. USA. – 1987. – Vol. 84 – P. 6919 – 6925. 19. Laboratory measurement of lipids, lipoproteins and apolipoproteins. Edited by N. Rifai, G. Russel Warnick. – 1994. 20. NCEP Expert Panel on Blood Cholesterol: Levels in children and adolescents: National Cholesterol Education Program (NCEP): highlights of the Report of the Expert Panel on Blood Cholesterol Levels in Children and Adolescents. Pediatrics 89:495–501, 1992 21. Obarzanek E, Kimm S, Barton B, Van Horn L, Kwiterovich P, Simons-Morton D, Hunsberger S, Lasser N, Robson A, Franklin F, Lauer R, Stevens V, Aronson Friendman L, Dorgan J, Greenlick M, on be-half of the DISC Collaborative Research Group: Long-term safety and efficacy of cholesterol-lowering diet in children with elevated low-density lipoprotein cholesterol: seven-year results of the Dietary Intervention Study in Children (DISC). Pediatrics 107:256–264, 2001 22. De Jongh S, Ose L, Szamosi T, Gagne C, Lambert M, Scott R, Perron P, Dobbelaere D, Saborio M, Tuohy MB, Stepanavage M, Sapre A, Gumbiner B, Mercuri M, van Trotsenburg AS, Bakker HD, Kastelein JJ, Simvastatin in Children Study Group: Efficacy and safety of statin therapy in children with familial hyper-cholesterolemia: a randomized, double-blind, placebo-controlled trial with simvastatin. Circulation 106:2231–2237, 2002 23. Starc TJ, Deckelbaum RJ Evaluation of Hypercholesterolemia in Childhood / Pediatrics in Re-view, 1996, Vol. 17, No. 3, pp. 94-97
01/20/2021 01:56:08 pm
Med-Apparatus
Med-Apparatus
Med-Apparatus medical devices and physiotherapy. Medical equipment bulletin board.
Russia
Med-Apparatus medical devices and physiotherapy. Medical equipment bulletin board.