Introduction
The leading cause of the development of atherosclerosis is currently considered to be a disorder of lipid metabolism and low-density lipoprotein (LDL) cholesterol (C) as the main indicator for assessing cardiovascular risk (CVR). Close to LDL is lipoprotein (a) (Lp(a)), the functional and pathophysiological significance of which has not yet been fully determined, despite the large number of original and review articles devoted to this issue since 1963, when the Lp(a) particle was first described by Norwegian professor of medical genetics Kare Berg.
Structure of Lp(a)
Lp(a) is an LDL particle containing an apolipoprotein A (apoA) molecule covalently linked to apolipoprotein B-100 (apoB) by a disulfide bond (Fig. 1) [1].
The uniqueness of the apoA protein lies in the fact that it is not found in any other class of lipoproteins and has a high degree of homology (up to 90%) to the kringle domain of plasminogen. The gene responsible for the synthesis of the apoA protein is localized in the long arm of chromosome 6, next to the plasminogen gene. However, no association has been demonstrated between high Lp(a) levels and the risk of venous thrombosis and thromboembolism [2]. Kringle domains are autonomous protein regions that fold into loops stabilized by disulfide bonds and play an important role in protein-protein interactions of the coagulation cascade, as well as in the binding of various mediators (interaction between proteins and phospholipids, biological membranes). There are ten types of kringle domains. Among them, only domain type 2 varies in copy number from 2 to 40, which determines the presence of multiple protein isoforms and heterogeneity of Lp(a) structure in the population [3]. ApoA is hydrophilic and can bind to damaged vascular endothelium, like plasminogen [4]. Lp(a) particles have a diameter of less than 70 nm and easily penetrate the endothelial barrier, where, like LDL, they accumulate and increase the risk of cardiovascular disease (CVD) [5].
Is a Lipoprotein(a) test necessary?
Why is it necessary to determine the level of Lipoprotein (a)?
Lipoprotein(a) consists of one molecule apo(a), apoB and low-density lipoproteins (LDL). A number of medical societies have recommended identifying high Lp(a) levels for the prevention of cardiovascular disease (CVD), and in June 2021, the Centers for Disease Control and Prevention approved the first ICD-10 codes to diagnose elevated Lp(a) levels.
There are currently no FDA-approved medications designed to lower Lp(a) levels. However, Lp(a) testing identifies people at increased risk of cardiovascular disease who have not been diagnosed by other tests, allowing them to receive timely treatment for other cardiovascular disease risk factors.
Limitations of various methods for determining Lp(A)
The most common methods for measuring Lp(a) are immunoassays, most of which use various polyclonal antibodies against apo(a), which react with the highly polymorphic krigling type IV type 2 domain. Given the high degree of size heterogeneity in apo(a), a polyclonal Lp(a) immunoassay may overestimate or underestimate the level of Lp(a) in human blood samples depending on the level of apo(a) in the test calibrators.
Another level of uncertainty relates to the expression of Lp(a) concentrations in mg/dL. Because Lp(a) particles are extremely variable not only in apo(a) size, but also in their lipid content and degree of glycosylation, it is incredibly difficult to determine the exact mass of Lp(a) in a reference standard and impossible to apply this value to secondary reference materials. to calibrators and ultimately to human samples.
In 2004, the World Health Organization/International Federation of Clinical Chemistry and Laboratory Medicine (WHO/IFCC) approved a reference reagent for Lp(a) immunoassays with precision specified in molar concentration (nmol/L). The monoclonal enzyme-linked immunosorbent assay (ELISA) method, independent of variations in apo(a) size, has been identified as the reference method, and studies have demonstrated comparability of Lp(a) results using the reference material (Clin Chem 2000; 46:1956-67) .
However, most commercially available immunoassays lack optimization to minimize the impact of apo(a) size variability, with the exception of one latex enhanced turbidimetric method that uses a mixture of apo(a) isoforms in its calibrators.
There are other analytical approaches for measuring Lp(a) that are less susceptible to apo(a) heterogeneity, such as ELISA methods that use a capture antibody recognizing apo(a) and a detection antibody against apoB, as well as methods based on electrophoretic separation of lipoprotein particles followed by apoB or cholesterol staining, followed by quantification.
What should laboratories consider when performing Lp(A) analysis?
Before performing Lp(a) studies, the laboratory must first assess the expectations of health care providers regarding the target patient population and the decision rules that define Lp(a) reference levels. Both physicians and laboratory technicians should be aware of racial/ethnic differences in Lp(a) levels. African Americans in particular tend to have higher levels of this particle than other racial/ethnic groups.
Laboratories should also consider the following questions about the analytical features of the Lp(a) test before making a selection: 1) Is the system WHO/IFCC certified for Lp(a) determination? 2) Is Lp(a) measured in nmol/L instead of mg/dL? Calculation using a conversion factor is not recommended. 3) Are the reagents insensitive to apo(a) size variability? Even if this is taken into account, there is a possibility of measurement error in patients with extreme apo(a) sizes. 4) And will patients have Lp(a) levels determined by different methods in several laboratories?
Expert opinion:
Thanks to advances in the standardization of methods for determining Lp(a) and more correct statistical processing of data, the position that elevated levels of Lp(a) is a risk factor for cardiovascular diseases, as well as peripheral vascular diseases, is considered clearly established and generally accepted. Diagnosis of elevated Lp(a) levels will allow timely initiation of treatment where it is really needed.
Metabolism and biological role of Lp(a)
Lp(a) is synthesized in the liver, where its catabolism also occurs. On the surface of hepatocytes there are many receptors involved in the process of endocytosis of Lp(a) molecules, including scavenger receptors B1 and fibrinogen receptors [3]. Along with the hepatic pathway of degradation, there is also a renal pathway. In a prospective study, it was found that the levels of apoA and apoB in hemodialysis patients were significantly lower than in healthy controls, while no difference in the rate of their synthesis was found [6]. The half-life of Lp(a) is longer than that of LDL and is 3.3 days.
Individual Lp(a) levels are approximately 90% determined genetically [7]. There is no single point of view on the physiological role of Lp(a), despite a large number of fundamental and clinical works on this topic. Africans are characterized by an increased concentration of Lp(a), which is on average 7 times higher than that of representatives of European and Asian populations [8]. Considering the high heterogeneity of the structure of this molecule, it can be assumed that the significance of Lp(a) is different not only in individuals of different races, but also in different populations of the same race. A.V. Tikhonov in 1980–1992. A comparative study of Lp(a) levels was carried out as part of a screening program for the unorganized population of one of the districts of Novosibirsk, indigenous residents of Chukotka and Gorny Altai. It was found that high levels of this lipoprotein are more typical for the indigenous population of Chukotka, living in the extreme climatic conditions of the Asian part of the continent. This may be due to the predominance in their diet of such food components as meat of animals and sea animals, and animal fats [8].
There is a hypothesis that suggests the participation of Lp(a) in tissue repair. It is assumed that the delivery of cholesterol to tissues in which there is an active repair process can occur through the connection of Lp(a) and fibrin. Due to the structural homology of the domains, apoA can also possess the qualities of various growth factors, including hepatocyte growth factor [9].
Lipoprotein structure (a)
Historically, the division of lipoproteins into different categories is based on the different densities of lipoprotein particles determined by ultracentrifugation:
- chylomicrons
- very low density lipoproteins (VLDL, VLDL)
- intermediate density lipoproteins (IDL)
- low density lipoproteins (LDL, LDL)
- high density lipoproteins (HDL, HDL)
- lipoproteins (a) (Lp (a))
The density of lipoprotein (a) (Lp(a)) is close to HDL. According to electrophoretic mobility - to VLDL. Structurally, Lp(a) particles are similar to LDL.
The lipoprotein(a) particle consists of cholesterol, triglycerides, Apo B, phospholipids and the apolipoprotein Apo(a). They have a similar lipid composition to LDL and, like LDL, contain 1 molecule of apo-B protein per particle. But the protein part of Lp(a) also contains a special protein, unique to these lipoproteins, the apolipoprotein Apo(a).
Apolipoprotein Apo(a) is a large hydrophilic and highly glycosylated protein that is similar in composition to plasminogen. Apolipoprotein Apo(a) consists of domains called “kringle” (pretzel, English), which, in fact, are similar to similar domains of plasminogen. Apolipoprotein Apo(a) consists of an inactive protease domain, one kringle V domain, and a variable number of kringle IV domains.
Different individuals in the gene encoding the apolipoprotein Apo(a) may have a different (from 12 to 51) number of DNA fragments encoding the apolipoprotein Apo(a) domain.
The number of “kringle” domains in Apo(a) is thus determined genetically and can vary from 12 to 51. As a result, significant polymorphism is observed in the population both in protein size and in the size of Lp(a) particles. And therefore, the molecular weight of the apolipoprotein Apo(a) protein in different individuals can range from ~280 to 800 kDa; There are currently 34 known lipoprotein(a) isoforms.
It is assumed that the apolipoprotein gene Apo(a) originated from repetitions of some parts of the plasminogen gene, the two genes being closely related to each other.
Apolipoprotein Apo(a) is synthesized in the liver and binds via a disulfide bond to newly synthesized apoB-100. Since both proteins interact with their C-terminal regions, apo B loses affinity for its receptor (LDL receptor). Lp(a) catabolism, unlike other lipoproteins, occurs in the kidneys and not in the liver.
In apolipoprotein Apo(a), the kringle domains are organized into a specific protein “motif” consisting of three loop-like structures stabilized by three disulfide bonds. This “motif” is also contained in a large number of proteins encoded by genes of the prothrombin family, including prothrombin, plasminogen, hepatocyte growth factor, urokinase, factor XII, tissue plasminogen activator.
Plasminogen is a precursor (proenzyme) of plasmin, the main enzyme that breaks down fibrin clots. It turned out that the size of apolipoprotein (a) determines the concentration of lipoprotein (a) in plasma. The smaller the size of the apolipoprotein Apo(a), i.e. the fewer “kringle IV” domains it contains, the higher the level of Lp(a) in the plasma and vice versa, the longer the apolipoprotein Apo(a) molecule, the lower the concentration of Lp(a).
In general, the level of apolipoprotein Apo(a) synthesis is determined by how quickly its isoforms are secreted. Smaller isoforms of the apolipoprotein Apo(a) are secreted more rapidly and therefore plasma Lp(a) levels are inversely proportional to the size of the apolipoprotein Apo(a).
Mechanisms of atherogenesis involving Lp(a)
It is likely that the atherogenicity of Lp(a) is multistage. The majority of Lp(a) proteins (35% of the pool) are represented by the apoB/apoA complex with a molecular weight ratio of 2:1. Due to the presence of such a specific structure, Lp(a), along with LDL cholesterol, can bind cholesterol and transfer it to the vascular wall [3]. In addition, under the influence of apoA, highly hydrophobic apoB acquires the ability to dissolve in water. The complex of apoA with apoB delays the degradation of apoB through the classical receptor pathway, thereby creating the prerequisites for its longer circulation in the blood plasma, modification changes and entry into cells through unregulated endocytosis.
In turn, Lp(a), easily penetrating the endothelial barrier [7], stimulates the expression of adhesion molecules by endothelial cells, and after oxidation inside the atherosclerotic plaque, it is phagocytosed by macrophages, transforming the latter into foam cells and releasing proinflammatory cytokines. Oxidized membrane phospholipids, which are mainly transported by Lp(a), are covalently bound to the apoA protein and provide additional pro-inflammatory potential to the plasma [10]. Additionally, Lp(a) can stimulate the proliferation of vascular smooth muscle cells and contribute to endothelial dysfunction. Both mechanisms are recognized as key in the development of the atherosclerotic process. The presence of Lp(a) and its specific components apoA in atherosclerotic plaques of the coronary arteries once again confirms its atherogenic qualities [3].
Cardiovascular risk
A high concentration of Lp(a) in the blood plasma is associated with an increased risk of developing atherosclerosis-associated diseases, but for most patients this indicator is a much weaker risk factor than the level of LDL cholesterol. According to European cardiologists and lipidologists, reflected in the latest recommendations for the diagnosis and treatment of dyslipidemia (2019), given the genetic determination of Lp(a) levels, its extremely high level (>180 mg/dl, or >430 nmol/l) may represent a new hereditary disorder of lipid metabolism, the prevalence of which is 2 times higher than heterozygous familial hypercholesterolemia, and the cardiovascular risk factors in such patients are identical. In this regard, it is recommended to measure Lp(a) levels at least once in every adult’s life. Lp(a) measurement is also justified (grade IIa, level of evidence C) in selected patients with a family history of premature CVD, and for the purpose of reclassifying those at borderline (moderate to high) risk, along with reclassifiers such as carotid atherosclerosis more than 25% and/or coronary calcification more than 100 units. Agatston [7].
Elevated Lp(a) levels may partly explain the residual risk in patients on adequate lipid-lowering therapy when LDL cholesterol targets are achieved [11]. A meta-analysis including data on cardiovascular outcomes of 29,069 patients, 14,536 of whom were treated with statins (AFCAPS, CARDS, 4D, JUPITER, LIPID, MIRACL and 4S studies), showed that even with statin therapy, when Lp(a) levels increase ) had a positive correlation with the composite endpoint (acute coronary syndrome, stroke, or revascularization) at three years of follow-up. Patients taking simva-, atorva-, or rosuvastatin and having an Lp(a) value ≥50 mg/dL had a 43% increase in CV risk compared with those with an adjusted Lp(a) value <15 mg/dL for other risk factors (age, gender, presence of CVD, diabetes mellitus, smoking, arterial hypertension (AH), LDL cholesterol, high density lipoprotein cholesterol (HDL)) [12]. A case-control study found that Lp(a) at a concentration of ≥50 mg/dL increased the risk of CVD by 2 times in young people (less than 45 years), by 3 times in people of the middle age group (45–60 years), however, it did not play a significant role in patients over 60 years of age, which creates theoretical prerequisites for including this indicator in multifactorial prediction models of cardiovascular outcomes in patients with atherosclerosis-associated diseases, which are now actively being developed [13, 14].
Author-compiler: Ph.D. biol. Sciences V.V. Velkov
Measuring the concentrations of Apo B and A1 - the key proteins of LDL-C and HDL-C - is the most accurate and unambiguous determination of the balance of pro-atherogenic and anti-atherogenic cholesterol, which assesses the risk of fatal and non-fatal myocardial infarction over the next five years. These extremely important provisions are based on the results of the AMORIS project, which was carried out in Sweden over a period of 8 years and 3 months. 98,722 men and 76,831 women were observed (age range from 20 to 80 years). During this period, 3,915 men and 2,461 women died from AMI and had significantly elevated Apo B levels and significantly reduced plasma Apo A1 concentrations. Thus, high levels of Apo B strongly correlate with an increased risk of CVD, and high levels of Apo A1 are a cardioprotective factor independent of gender. Moreover, Apo B is a stronger indicator of CVD risk than LDL-C, especially when LDL-C is normal or reduced.
The Apo B/Apo A ratio indicates the risk of CVD regardless of the level of cholesterol-related lipids, and even when the level of these lipids is normal.
In this case, the assessment of CVD risk can be expressed in one number (see Fig. 3). It is significant that the Apo B/Apo A1 ratio has a stronger association with CVD risk than the TC/LDL-C or LDL-C/HDL-C ratios.
Rice. 3. The risk of developing MI depending on the ratio of Apo B and Apo A1 concentrations
Overall, the results of this and other similar prospective studies clearly indicate that:
- the risk of atherosclerosis is associated not so much with the concentration of cholesterol, but with the number of circulating atherogenic particles that easily bind to the walls of blood vessels and quickly penetrate into the arterial walls,
- Apo B is the most accurate indicator of CVD risk,
- Apo B is the most adequate indicator of the effectiveness of lipid-lowering therapy,
- it is necessary to replace the determination of total cholesterol and LDL-C with measuring the concentration of Apo B,
- Apo B/Apo A1 ratios are superior to the indicator properties of all other cholesterol ratios in terms of their accuracy in assessing coronary risks in patients with asymptomatic CVD and in people with diabetes.
Of course, the determination of total cholesterol, LDL-C and HDL-C has by no means lost its diagnostic value. But it should be kept in mind that according to well-established research, abnormal levels of these markers do indicate the presence of atherosclerosis, but their normal levels do not necessarily indicate that atherosclerosis does not exist.
LIPOPROTEIN (a) – PREDICTOR OF GENETIC PRESPOSITION TO CVD Lp(a) [Lp(a)], or “small lipoprotein a” is LDL-C with an “appendage” – Apo(a), a large glycoprotein, which, through a disulfide bond covalently bound to apolipoprotein Apo B, which is part of LDL-C. Lp(a) synthesis occurs in the liver, but Lp(a) catabolism, unlike the catabolism of other lipoproteins, occurs in the kidneys and not the liver. Apo(a) has homology to human plasminogen and consists of domains called “kringle”, which are similar to those of plasminogen. Different individuals in the gene encoding Apo(a) may have a different (from 12 to 51) number of DNA fragments encoding “kringle” domains. As a result, significant polymorphism is observed in the population in terms of protein size and particle size of LP(a).
As has been established, the smaller the size of Apo(a), i.e. the fewer “kringle” domains it contains, the higher the level of Lp(a) in the plasma, and vice versa, the longer the Apo(a) molecule, the lower the concentration of Lp(a).
The level of Lp(a) in plasma is more than 90% determined genetically and depends mainly on the rate of Apo(a) biosynthesis, which is inversely proportional to the size of Apo(a). Beginning in early childhood, Lp(a) concentrations increase, reach a plateau in adulthood, and then remain virtually unchanged. A further increase in Lp(a) levels is observed only in postmenopausal women. Unlike most lipid risk factors, the risk associated with elevated Lp(a) levels is independent of age, sex, diet, and living conditions. However, since Lp(a) catabolism occurs in the kidneys, renal pathologies increase Lp(a) levels due to decreased catabolism of its particles. The normal physiological role of Lp(a) is unclear. It is believed that Lp(a) is either somehow involved in the metabolism of cholesterol and triglycerides (since it is similar to LDL-C), or takes some part in coagulation processes, since Apo(a) is similar to plasminogen. Elevated levels of Lp(a) cause CVD disease due to the proatherogenic nature inherent in LDL-C and stimulate thrombus formation due to the prothrombotic properties of apolipoprotein Apo(a).
Elevated levels of lipid lipid (a) are the most common genetically mediated disorder of lipid metabolism in individuals with early CVD. In general, elevated Lp(a) concentrations increase coronary risk, particularly in men with high LDL-C and low HDL-C levels. Moreover, measuring Lp(a) levels allows one to determine the risk of ischemic stroke independent of other risk factors. Measuring Lp(a) levels in renal pathologies, as well as before and after hemodialysis, allows us to assess the risk of subsequent vascular events. In type 1 diabetes, Lp(a) levels above 30 mg/dL are associated with a doubled risk of vascular complications, including coronary artery disease, peripheral artery disease, and cerebrovascular disease. In type 2 diabetes, elevated levels of Lp(a) are also a predictor of CVD.
LP(a) is a risk factor and predictor of genetic predisposition to cardiovascular and microvascular diseases and genetically mediated ischemic strokes. Lp(a) levels should be measured in patients:
- with early cases of CVD,
- in those with a family history of frequent cases of CVD (suspicion of genetic predisposition),
- with a diagnosis of CVD, but without traditional risk factors,
- in whom hypercholesterolemia does not decrease with statin therapy,
- with renal diseases,
- with diabetes types 1 and 2.
Important information When direct determination of LDL-C and immunoturbidimetric measurement of Apo B, the measurement results always include the concentrations of Lp(a) and Apo(a). When calculating LDL-C determination, the result also includes Lp(a) concentrations. Therefore, at high levels of LDL-C and Apo B, it is advisable to determine what contribution to them is made by a genetically mediated increase in the concentration of highly atherogenic and prothrombotic Lp(a) and Apo(a).
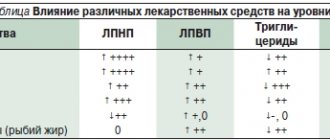
Lipid-lowering therapy and Lp(a)
Lp(a) levels are under strict genetic control, change little throughout life and do not decrease significantly when following a diet and taking conventional lipid-lowering drugs. Although an association has been found between Lp(a) levels and increased CV risk, there is still a lack of evidence to suggest that lowering Lp(a) levels improves prognosis [3]. Randomized trials of niacin and cholesteryl ester transfer protein (CETP) inhibitors, which reduce Lp(a) concentrations by 20–30%, have not provided evidence that its reduction provides a reduction in the risk of developing atherosclerosis-associated diseases beyond that expected with a marked decrease in the level of lipoproteins containing apoB [15].
At the same time, recent data regarding PCSK9 inhibitors indicate a decrease in Lp(a) levels by 20–30% during therapy with these drugs, which may play a role in the integral reduction in CV risk associated with cumabs [16]. It should be noted that patients with mutations causing a decrease in the number of LDL receptors may not respond to cumab therapy [17].
Another drug is mipomersen, an antisense oligonucleotide, whose action is aimed at reducing the synthesis of apoB-100-containing lipids - LDL, Lp(a) - through mRNA. Although mipomersen was approved by the US Food and Drug Administration in 2013 as adjunctive therapy for familial hypercholesterolemia in patients over 12 years of age, the European Medicines Agency refused to authorize the drug that same year due to concerns safety, because it caused adverse reactions (local injection reactions, liver steatosis, increased transaminase levels, flu-like reactions). Mipomersen reduces LDL levels by 26.4%, Lp(a) by 22.7% [18].
Another direction that ensures plasma reduction of Lp(a) is IONIS-APO(a)Rx, an antisense oligonucleotide molecule that selectively interacts with apoA mRNA. RNase H1 recognizes the RNA-DNA duplex formed when ISIS-APO(a)Rx binds to the complementary sequence of apoA mRNA and cleaves the target, thereby reducing apoA synthesis and preventing the formation of Lp(a) (Fig. 2) [19]. In a phase 1 study in healthy volunteers with Lp(a) levels ≥25 nmol/L, IONIS-APO(a)Rx dose-dependently reduced Lp(a) levels, with a maximum dose of 300 mg achieving a reduction of 77.8 % [15].
Currently, lipid apheresis is the only method to significantly reduce plasma Lp(a) levels (>60% in one procedure) that has shown effectiveness in observational studies, but this type of therapy is invasive and expensive, which limits its use in clinical practice [3] . Data from the German Lipid Apheresis Registry show that regular use of the procedure in patients with high levels of Lp(a) and LDL cholesterol during lipid-correcting therapy reduces the risk of cardiovascular events [15].
The following is a clinical observation demonstrating the rarity of determining the Lp(a) content even in the conditions of a “non-classical”, progressive course of CVD and actualizes the need to include it in the standard lipid examination at least once during the life of any adult according to the European and latest Russian recommendations “Diagnostics and correction of lipid metabolism disorders for the purpose of prevention and treatment of atherosclerosis” [20].
Clinical observation
Patient B., 62 years old. In 2003, at the age of 45, she suffered a transient ischemic attack, at which time stage II hypertension was first detected. In the lipid profile: total cholesterol 6.1 mmol/l, LDL cholesterol 4.2 mmol/l, HDL cholesterol 1.0 mmol/l, triglycerides 2.2 mmol/l. Blood glucose level was 5.1 mmol/l, glomerular filtration rate (calculated using the CKD-EPI formula) 98 ml/min/1.73 m2, Lp(a) content was not determined.
She has no family history, does not smoke, and has a regular menstrual cycle. Upon examination, no clinically significant pathology was detected; body mass index was 28 kg/m2. Along with acetylsalicylic acid, the patient was prescribed three-component antihypertensive therapy, including amlodipine, losartan and hydrochlorothiazide, and target blood pressure values were achieved. Taking rosuvastatin 10 mg/day allowed us to achieve the desired values of the lipid spectrum - LDL cholesterol 2.4 mmol/l (the target LDL cholesterol value at that time, according to current clinical recommendations, was <2.5 mmol/l). Prediabetes has been registered since the age of 49, and he takes a long-acting form of metformin at a dose of 1500 mg/day.
Despite high adherence to drug therapy, the patient, aged 54 years, manifested coronary heart disease (CHD) in the form of stable angina pectoris of functional class II (FC), decreased tolerance to physical activity. At the age of 56, due to frequent cervicalgia, which was disturbing without connection with an increase in blood pressure, magnetic resonance imaging of the brain was performed and multiple small vascular lesions measuring 1.5–2.5 mm were detected, signs of a previous right hemisphere cerebral infarction: lesions measuring 15 ×20 mm in the border blood supply basin of the middle cerebral artery and anterior cerebral artery. Lipid profile: LDL cholesterol 2.2 mmol/l, triglycerides 1.47 mmol/l.
In November 2021, at the age of 61, she was undergoing a routine examination with a diagnosis of coronary artery disease. Angina pectoris II FC. Hypertension stage III, hypertension stage III, risk 4. Chronic heart failure stage I (NYHA FC II). Chronic cerebral ischemia, residual phenomena of acute cerebrovascular accident in the right hemisphere in the border blood supply basin of the middle cerebral artery, anterior cerebral artery. Marked increase in Lp(a) content. Abdominal obesity II degree. Impaired glucose tolerance.
The patient underwent duplex scanning of the brachiocephalic arteries: stenosis of the right subclavian artery 20–25%; stenosis at the border of the middle and distal third of the left common carotid artery 20–25% due to a heterogeneous atherosclerotic plaque; prolonged stenosis of 25% in the middle third of the left common carotid artery due to a heterogeneous atherosclerotic plaque. Holter monitoring of electrocardiography was performed: against the background of sinus rhythm with a heart rate of 135 per minute, a single episode of descending depression of the ST segment was recorded in lead V3–V5 with an amplitude of 0.1 mV. According to echocardiography: signs of left ventricular myocardial hypertrophy, diastolic dysfunction, thickening of the aortic walls, left ventricular ejection fraction - 62%.
Complete blood count and complete urinalysis without clinically significant changes; glomerular filtration rate 92 ml/min/1.73 m2, glycated hemoglobin 5.6%, thyroid-stimulating hormone 3.5 mIU/l. During therapy with rosuvastatin 20 mg/day in combination with ezetemibe 10 mg/day: LDL cholesterol 1.4 mmol/l, HDL cholesterol 1.1 mmol/l, triglycerides 1.7 mmol/l. The first measurement of Lp(a) level showed a value of 440.25 nmol/l (with reference values up to 50 nmol/l), which indicates a hereditarily determined risk of CVD, comparable to the risk of heterozygous familial hypercholesterolemia. However, if in the latter case the timing of initiation of lipid-correcting therapy is clearly defined - in adults from the moment of diagnosis, and in children from 8 years of age [10], then with a significant increase in Lp(a) content, the start time, tactics of drug intervention, target lipid values within primary and secondary prevention are unknown. Currently, the patient continues to take previously selected therapy, including combined lipid-lowering therapy (rosuvastatin 20 mg/day in combination with ezetemibe at a standard dose); she refused the proposed replacement of ezetemibe with a PCSK9 inhibitor for financial reasons.
HDL cholesterol test (alpha cholesterol, HDL)
Alpha cholesterol plays a large role in the disposal of excess cholesterol molecules. High-density lipoproteins circulate in the bloodstream and, where there is an increased concentration of cholesterol unused by cells, bind it and transport it to the liver. In hepatocytes, HDL is released from cholesterol under the influence of liver enzymes, and the latter is excreted from the body with bile.
The higher the level of HDL cholesterol in the blood, the more actively excess cholesterol is removed from tissues, the less likely the formation of atherosclerotic plaques on the vascular walls, and the less likely it is to develop cardiovascular pathology. Thus, HDL cholesterol is “good” or antiatherogenic.
HDL testing is mandatory when studying lipid metabolism. This analysis is also prescribed as a marker of the effectiveness of non-drug and pharmacological correction of metabolism. The norm is at least 1.5 mmol/l. The lowest value is 1.0.
All patients who have any of the following factors are required to monitor this indicator:
- Age over 40 years;
- Sedentary or sedentary lifestyle;
- The predominance of animal fats, refined and simple carbohydrates in the diet;
- Smoking;
- Frequent stress;
- Alcohol abuse;
- Having excess body weight;
- Cardiovascular diseases in close relatives;
- Jumps in blood pressure, especially frequent increases;
- Borderline values of glucose and lipid metabolism in a biochemical blood sample.
The following also leads to a pathological decrease:
- Kidney failure;
- Genetic defects in the synthesis of lipoprotein components;
- Hepatic pathology with chronic stagnation of bile (cholelithiasis, bile duct dyskinesia, hepatitis, cirrhosis, toxic liver damage).
Long-term HDL deficiency leads to the accumulation of cholesterol in the blood vessels and tissues of internal organs, significantly increasing the risk of:
- Atherosclerosis of the aorta and arteries;
- Atherosclerotic cardiosclerosis;
- Angina pectoris;
- Heart rhythm disturbances;
- Myocardial infarction;
- Stroke;
- Renal dysfunction;
- Circulatory disorders in the intestines and limbs.
To prevent these diseases, it is necessary to regularly monitor HDL cholesterol levels, especially if there are risk factors.