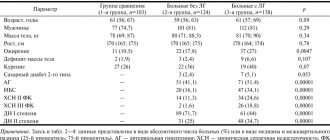
Cardiomyopathy can come in various forms, among which left ventricular hypertrabecularity is considered the most rare. The disease is mainly identified in children, although it sometimes occurs among adults. In severe cases, even a heart transplant may be required.
Left ventricular hypertrabecularity (LVHT, non-compact myocardium, spongiform cardiomyopathy) is a condition in which the muscular wall of the left ventricle of the heart becomes “spongy”. This can affect the heart's ability to pump efficiently and the electrical conductivity of the myocardium.
Left ventricular (LV) hypertrabecularity cannot be completely cured, but medication can help patients reduce symptoms.
The disease occurs rarely and is considered a congenital pathology. During the embryological development of the heart, two layers are formed: normal and pathological (spongy). The development of the clinic and further examination of the patient makes it possible to make a diagnosis and prescribe symptomatic treatment.
Video: Noncompaction myocardium (NCM) - Non-compact (“spongy”) myocardium (NCM) or spongy cardiomyopathy
Introduction
One of the rare and difficult to diagnose, predominantly congenital, primary cardiomyopathies is non-compact, or spongy, myocardium of the left ventricle, which is manifested by hypertrophy of the left ventricular myocardium, its excessive trabeculation and the formation of wide intertrabecular spaces due to disruption of the intrauterine process of myocardial compaction. Diagnosis of non-compact myocardium of the left ventricle is difficult due to the lack of typical clinical signs of the disease and lack of knowledge.
History of disease detection
The first information about non-compact myocardium of the left ventricle dates back to 1932, when S. Bellet, during an autopsy of a newborn with aortic atresia and coronary ventricular fistula, revealed a spongy structure of the myocardium. In the 60–70s of the last century, publications began to appear in foreign literature that described changes in the myocardium in the form of increased trabecularity in combination with various heart pathologies: obstruction of the outflow tract of the left and right ventricles, combined cyanotic congenital heart disease, atrial septal defect, interventricular septum, pulmonary stenosis, anomaly of the coronary arteries. However, this pathology is defined as non-isolated non-compact ventricular myocardium.
It was later shown that non-compact left ventricular myocardium can also occur in combination with neuromuscular diseases such as metabolic myopathy, Barth syndrome, Ohtahara syndrome and others. The literature describes cases of non-compact myocardium in hereditary defects of the facial skull - protruding forehead, bilateral strabismus, micrognathia, cleft palate, etc.
In 1984, an isolated pathology in the form of a sinusoid in the left ventricle was described. A year later, the authors described a case of diagnosed atypical dilated cardiomyopathy in a 21-year-old patient who was diagnosed with acute left ventricular failure at the age of 15 years. Echocardiographic examination revealed channel-like structures in the thickened and hypokinetic left ventricular myocardium; angiography revealed a “honeycomb” structure of the left ventricular wall. A year later, the same authors described a similar clinical case in a 21-year-old student during an autopsy with pronounced sinusoids in the hypertrophied myocardium. In 1990, the results of observation of 8 cases of non-compact myocardium in children were presented. Despite the fact that the number of publications on cases of non-compact myocardium of the left ventricle in the available literature is increasing, there is not enough information about isolated non-compact myocardium in children [1, 5, 6, 8, 10].
Description
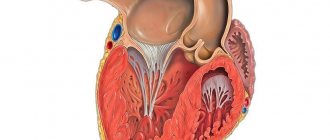
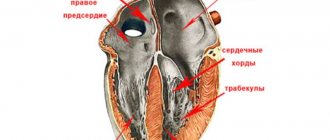
Left ventricular hypertrabecularity is a heart condition in which the walls of the left ventricle (the lower chamber of the left side of the heart) are not as thick as they normally should be. This leads to the formation of channels called trabeculae in the heart muscle. The left ventricle takes on a characteristic “spongy” appearance, a bit like a honeycomb. Although the left ventricle is primarily affected, some cases also show hypertrabecularity of the right ventricle.
The first description of the disease dates back to 1926 after examining a sick child.
LVHT is described by the American Heart Association as a type of genetic cardiomyopathy (a disease of the heart muscle caused by a change in a person's genetic makeup). However, the World Health Organization (WHO) describes it as an “unclassified” type of cardiomyopathy. The pathology may also be associated with dilated or hypertrophic cardiomyopathy. There are currently no universally agreed upon diagnostic criteria for the condition.
Pathology can develop at any age. In some cases, a combination of LVH with other congenital diseases is determined, in others an isolated form of the disease develops.
Epidemiology
The prevalence of non-compact left ventricular myocardium is, according to some authors, 0.05–0.24%, others – 0.014–0.14% in the general population. It can be assumed that due to the advent of the latest modern diagnostic methods, the detectability, and therefore the prevalence, of this pathology may increase. It is known that non-compact myocardium is more common in male patients than in female patients, ranging from 56 to 82% [8, 9].
The authors provide a link to data from 2003 on an epidemiological study of primary cardiomyopathies in Australian children, where non-compact myocardium was identified in 9.2% of cases of the total number of primary cardiomyopathies and was in third place after dilated and hypertrophic cardiomyopathies [1].
Causes and prevalence
LVH is basically a genetic disorder caused by an altered or “mutated” gene. If the disease is genetic, it can be inherited (passed from parent to child) and is therefore often common in certain families. For this reason, it is recommended to screen first-degree family members (parents and siblings) if someone close to them has previously been diagnosed with LVHT.
The prevalence of LVH is not really known, although some have suggested that the disease affects men more often than women. Nowadays, more people are diagnosed with LVH because cardiography is now more advanced, so non-consolidated areas can be seen and diagnosed quite accurately.
LVHT is sometimes diagnosed in patients with dilated or hypertrophic cardiomyopathy.
Some statistics:
- LVHT is registered in 80% of cases in men.
- In children, LVH is most common, as it ranks third after hypertrophic and dilated forms of cardiomyopathy.
- Among diagnosed cardiomyopathies, LVHT is detected in 9.2% of cases.
- The total share of pathology accounts for 0.014% of cases (according to E. Oechslin).
Pathogenesis
Disturbances in embryogenesis occur in the early stages. Normally, by the 26th day of intrauterine development, the myocardium is represented by a complex sponge-like structure of muscle trabeculae with multiple intertrabecular pockets - lacunae. This structure is a necessary condition for normal development, since in this period the coronary vessels have not yet been formed and young cardiomyocytes are forced to consume oxygen directly from the chambers of the heart. By the 31st day, the walls of the heart become denser, the lacunae close and partially participate in the formation of coronary vessels. If compaction of myocardial fibers does not occur for any reason, then the child is born with a rare anomaly - non-compact (spongy) myocardium. The main symptom of this disease is deep trabeculae in the myocardium of the left ventricle and the interventricular septum, which entails a decrease in systolic function of the left ventricle. Complete non-compaction of the myocardium occurs in children and is usually combined with other congenital anomalies, most often stenoses of the outflow tract of the right and left ventricles. The mortality rate among children with concomitant pathology of complete non-compact myocardium is very high. Adults with isolated spongy myocardium usually have no complaints; this pathology can be detected accidentally during examination due to other conditions.
It has been established that in a healthy child the left ventricle has up to three visible trabeculae and is less trabecular compared to the right ventricle. In the case of intrauterine disturbance of endomyocardial morphogenesis, processes of disorganization of myocardial compaction occur. During embryogenesis, the depressions interact with the ventricular endocardium and, with further compaction of the myocardium, are transformed into capillaries. Non-compact myocardium is represented by the presence of more than three deep intertrabecular depressions in hypertrophied segments of the left ventricular myocardium. In addition, the ventricles are often hypokinetic. It was found that the deep intertrabecular recesses are covered with epithelium, which indicates a relationship with the ventricular endocardium. In contrast to established disorders in the left ventricle, it is not possible to differentiate increased trabecularity of the right ventricle, since the right ventricle normally has increased trabecularity. However, the etiopathogenetic mechanisms of the development of the disease have not yet been fully studied [1, 2, 9].
Genetic aspects
The literature contains data on both sporadic forms of the disease and familial cases of isolated non-compact myocardium of the left ventricle. Isolated non-compact left ventricular myocardium is a genetically heterogeneous disease. The authors distinguish several types of left ventricular non-compact myocardium syndrome: type 1 - thickening of the left ventricular wall and interventricular septum without left ventricular dilatation is observed, type 2 - a combination of signs of left ventricular non-compact myocardium and dilated cardiomyopathy is observed, and X-linked type.
Type 1 syndrome of non-compact left ventricular myocardium is inherited in an autosomal dominant manner, in some cases it is the result of new mutations. In this type, both isolated syndrome of non-compact left ventricular myocardium and its combination with heart defects are observed. The most common defects are atrial septal defects, isolated or multiple ventricular septal defects, pulmonary artery stenosis and other defects. The cause of this disease is mutations in the dystrobrevin gene, which is located at the 18q12.1-q12.2 locus.
Type 2 is inherited in an autosomal dominant manner. There is no combination of left ventricular non-compact myocardium syndrome with heart defects. The gene responsible for this disease is still not known, but the 11p15 locus linked to this disease has been mapped. Therefore, direct diagnosis of this disease is not yet possible.
The X-linked type is inherited as an X-linked recessive type. This rare hereditary disease develops as a result of mutations in the G 4.5 (TAZ) gene, which is located on the X chromosome in the Xq28 region. This gene encodes the taffazin protein, which is an essential structural component of the membranes of skeletal and cardiac muscles, and also takes part in myocardial morphogenesis. Barth syndrome, other X-linked infantile cardiomyopathies, and X-linked endocardial fibroelastosis are associated with this gene. Although this mutation was not detected in adult patients, which may indicate other causes of the disease. Direct DNA diagnosis of this type of disease can confirm the presence of the disease. Women can be carriers of a mutant gene; they have a 50% chance of passing on the gene carrying the mutation to their sons. All daughters of female carriers will be healthy, but half of them will also be carriers of the mutant gene. However, not every mother who gives birth to a boy with left ventricular non-compact myocardium syndrome is diagnosed with a mutant gene. This fact can be explained by the fact that the mutation occurred for the first time in the patient. To determine the carriage of a mutant gene, molecular genetic examination methods are necessary [7].
Development mechanism
LVT - this pathology usually forms when the child is still an embryo developing in the womb. This means that a person is born with this disease. However, in some people the condition can develop after birth and is called “acquired” LVH.
The heart is made up of a thick layer of muscle cells (called the myocardium). Normally, muscle cells in the myocardium are tightly connected to each other and are therefore considered “compacted”.
During the development of the embryonic heart, the cardiac muscle fibers are initially “unconsolidated” and unable to contract normally. This is necessary to form a thick muscle wall. LVH occurs when this part of the heart's normal development (called the “remodeling phase”) is interrupted, causing part of the myocardium to remain loose rather than compact.
Classification
Since 1995, at the proposal of the WHO, according to the classification of cardiomyopathies, non-compact myocardium has been classified as a group of unclassified cardiomyopathies. In accordance with the classification of cardiomyopathies proposed by the American Heart Association (AHA, 2006), non-compact left ventricular myocardium belongs to primary genetic cardiomyopathies (unclassified cardiomyopathies). Since 2008, the European Society of Cardiology has proposed a classification of known and rare cardiomyopathies. In accordance with this classification, two large groups of cardiomyopathies are distinguished: familial and non-familial. The familial form is diagnosed when the disease is present in more than one family member or when the same genetic mutation occurs in cases of diseases in the family. The familial form of cardiomyopathy is often a monogenic disease. Of the congenital nonfamilial cardiomyopathies, only idiopathic ones were included in the classification. Acquired non-familial forms of cardiomyopathies are not included in this classification, since acquired cardiomyopathies have significant differences in diagnosis and treatment due to the fact that they are a complication of the disease, and not its clinical sign [3, 7–9].
Clinical signs
The main symptoms of non-compact myocardium are manifestations of heart failure, arrhythmias (ventricular tachycardia, atrial fibrillation, Wolff-Parkinson-White syndrome), thromboembolism (pulmonary embolism, blood clots in the ventricles of the heart), especially pronounced in patients with reduced left ventricular function. In addition to cardiac symptoms, facial dysmorphism was often observed in children. In adults, complete block of the bundle branches is most often observed. Also, children were more likely to have a familial type of the disease than adults. A constant sign of the disease in children and adults was the presence of non-compact myocardial segments. The severity of the condition of patients of both age groups was determined by the degree of myocardial non-compaction [1–3, 8, 9].
Diagnostics
To diagnose non-compact myocardium of the left ventricle, various methods of instrumental examination are used: echocardiography, magnetic resonance imaging, less often - computed tomography, positron emission tomography.
To visualize the cavity of the left ventricle, echocardiography is used, often two-dimensional, as well as three-dimensional. It is especially important to use echocardiography when conducting family screening in order to identify patients in the absence of complaints and symptoms of the disease.
The main echocardiographic sign of non-compact myocardium is the presence of increased trabecularity of the left ventricle. However, increased trabecularity may be a normal variant, but with non-compact myocardium, it can most often be determined between the anterolateral wall and the interventricular septum. For a more accurate diagnosis of non-compact myocardium, several echocardiographic studies are recommended, since the existing pathological trabecularity can be visually smoothed out when obtaining flat sections. It is also recommended to use parasternal short-axis imaging at end systole for differential diagnosis.
There are several options for diagnostic criteria for isolated non-compact left ventricular myocardium.
Criteria for diagnosing isolated non-compact myocardium of the left ventricle according to TK Chin (1990): absence of any other concomitant structural abnormalities of the heart; parasternal long axis, subcostal and apical views; determining the depth of excavations; measured at the end of diastole, the ratio between the distances from the surface of the epicardium to the beginning of the notches and from the epicardium to the end of the trabeculae is ≤ 0.5; increased trabecularity and deep intertrabecular depressions.
Criteria for the diagnosis of isolated non-compact myocardium of the left ventricle according to R. Jenni (1999): absence of concomitant cardiac anomalies; visualization at the level of the parasternal short-axis and apical projection reveals a typical two-layer myocardial structure with a thin compact outer (epicardial) layer and a thicker non-compact inner layer (endocardial) layer, which consists of a trabecular mesh structure with recesses deep to the endocardium (the maximum end-systolic ratio of the non-compact endocardial layer and compact myocardium is 2); segmental localization of abnormal myocardium predominates (that is, non-compact myocardium dominates at the apex (about 80%) and in the middle sections of the lower and lateral walls of the ventricle); using color Doppler, deep perfusion of intertrabecular grooves can be detected (unlike myocardial sinusoids, intertrabecular grooves do not communicate with the coronary blood flow).
In 2006, S. Lilje and co-authors proposed a method for quantitatively assessing the degree of myocardial non-compaction depending on the values of the above ratio: a ratio value of 0.33–0.26 corresponds to “mild” non-compaction, 0.25–0.2 – moderate, less than 0 ,2 - heavy. This value is recognized as a prognostic sign of the further development of the disease and the occurrence of complications, since it closely correlates with the degree and rate of development of heart failure.
Authors C. Stollbeger and J. Finsterer believe that to make a diagnosis of non-compact myocardium of the left ventricle, the following conditions are necessary: the presence of more than three trabeculae extending from the wall of the left ventricle in the apex to the papillary muscle and visualized in at least one position; perfusion of the intertrabecular spaces of their ventricular cavity, recorded by Doppler echocardiography.
The above criteria for diagnosing isolated non-compact myocardium of the left ventricle are echocardiographic signs confirming the diagnosis.
It has been established that most often (about 80%) the apical and middle segments of the lower and lateral walls of the left ventricle are affected.
The magnetic resonance imaging method for diagnosing isolated non-compact myocardium of the left ventricle is the most informative, especially when using three-dimensional visualization. The computed tomography method is less commonly used. Both of these methods have high resolution and are considered very promising in the diagnosis of non-compact myocardium of the left ventricle, although the results of their use are not sufficiently described in the literature [4–6, 8, 10].
Video
More photos Author(s):
L.V. Krasheninnikov, veterinary cardiologist, Innovative Veterinary Center of the Moscow Veterinary Academy named after. K.I. Skryabina [email protected]
Journal:
No. 6-2017
Key words
: non-compact myocardium of the left ventricle (NCLV), hypertrabecularity, heart failure
Kew words
: noncompaction cardiomyopathy (NCCMP), hypertrabecularity, heartfailure
Abstract
Non-compact myocardium of the left ventricle is very rare a form of congenital cardiomyopathy characterized by hypertrabecularity of the left ventricular myocardium.
During embryogenesis, with non-compact (hypertrabecular) left ventricular myocardium (LVLV), two layers of cardiac muscle are formed - normal compact and pathological non-compact. Clinically, this type of cardiomyopathy is manifested by ventricular arrhythmias, heart failure, thromboembolism, sudden death, and can also be asymptomatic. The main diagnostic methods are echocardiography, computed tomography and magnetic resonance imaging. Summary
Left ventricular noncompaction is a very rare form of genetic cardiomyopathy, that is characterized by hypertrabecularity of left ventricular myocardium.
During embryogenesis in case of Noncompaction cardiomyopathy (NCCMP) two layers of cardiac myocardium are formed – normal compact and pathological non compact. Clinically this type of cardiomyopathy presents with ventricular arrhythmias, heart failure, thromboembolic events, sudden death and also can be asymptomatic. Echocardiography, CT and MRI can be helpful in providing diagnosis of NCCMP. Disease Description
Non-compact (hypertrabecular) left ventricular myocardium (LVLM), also formerly known as spongy myocardium, is one of the rare primary cardiomyopathies characterized by prominent left ventricular trabeculae and deep intertrabecular recesses. In this case, two layers are formed - non-compact with reduced contractile function and compact. In humans, LVNC occurs at any age and can be isolated or combined with other congenital heart diseases.
The disease was first described in a child in 1926 and has long been considered a type of apical hypertrophic cardiomyopathy (HCM). Currently, LVNC is classified as an unclassified cardiomyopathy (European classification).
Etiology, genetics and embryogenesis
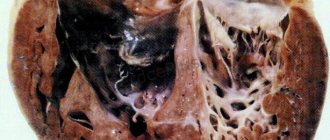
The cardiac muscle develops from the myoepicardial plate of mesoderm in the first trimester of pregnancy, while initially the myocardium is represented by a group of separate fibers separated from each other by wide sinusoids. During embryogenesis, the fibers gradually become denser and the intertrabecular spaces become narrower. The process proceeds from the base of the heart to its apex and from the epicardium to the endocardium. Disruption of this process leads to the fact that zones of undensified, “non-compact” trabeculae remain, separated by deep intertrabecular recesses (Fig. 1).
To date, the etiology of the disease remains poorly understood. There is evidence of the non-heritable and inherited nature of the disease, predominantly autosomal dominant and X-linked. The development of LVNC in humans may involve various mutations of genes, including those encoding the synthesis of sarcomeres - MYH7, ACTC, TNNT2, those responsible for the development of HCM - MYBPC3, proteins involved in the organization of the cytoskeleton - LDB3, Lamin A/C, cardiac-specific - CSX , alpha-dystrobrevin - Cypher/ZASP, dystropin and some others.
In particular, an interesting common R820W mutation of the MYBPC3 gene has been identified, causing HCM in Rag Doll cats, in homozygous people - LVML and HCM, and in heterozygous people the expression is minimal. Prevalence and classification
Currently, the prevalence of the disease in the human population is greatly underestimated due to the difficulties of diagnosis. This pathology is often described as other forms of cardiomyopathies, blood clots, tumors, etc.
The disease is more often registered in men - up to 80% of cases according to various authors. In pediatric practice, LV NML ranks third after HCM and DCM, accounting, according to some data, about 9.2% of all diagnosed cardiomyopathies. The overall proportion of the disease is about 0.014% (E. Oechslin). To date, one case of biventricular NML without hypertrophy has been described and histologically confirmed in a Maine Coon with the A31P mutation of protein C, responsible for HCM in this cat breed, and the animal was observed every 6 months for 6 years until death.
LVNC can be either isolated or combined with other cardiomyopathies, neuropathies and congenital heart defects. In humans, it most often occurs together with ventricular or atrial septal defects, but can also be combined with other congenital heart defects (CHD).
Pathogenesis and clinical picture
Disruption of the normal architecture of the cardiac muscle, manifested in the form of two layers, compact with normal contractility and non-compact with reduced, leads to a decrease in the overall contractility of the ventricular myocardium, and disturbance of microcirculation leads to chronic ischemia. These two factors, as well as the size of the non-compact part of the heart muscle in relation to the compact one, determine the severity and rate of development of chronic heart failure in the patient, both systole and diastole suffer, which in some cases can be of a restrictive type.
Clinically, LVNC manifests itself as chronic heart failure, less commonly, ventricular and supraventricular arrhythmias, an increased likelihood of thrombus formation, or is asymptomatic.
The development of chronic heart failure is associated with systolic and diastolic dysfunction and the resulting cardiomegaly and retrograde congestion in the corresponding circulatory system.
Due to pronounced structural changes in the myocardium, arrhythmias are a pathognomonic syndrome for LVNC and may be the only manifestation of the pathology. The most common ventricular arrhythmias are ventricular tachycardia and extrasystole, and the mechanisms responsible for their development are presumably similar to those in arrhythmogenic right ventricular cardiomyopathy. Other common disorders include atrial fibrillation, AV block, and bundle branch block due to progressive endomyocardial fibrosis. WPW syndrome is less common, and the additional pathway is often localized in the anteroseptal segment in the area of the fibrous ring of the tricuspid valve. Accessory pathways are the main cause of sudden death in LVNC.
The increased likelihood of blood clots depends on several reasons. On the one hand, these are increased sizes of the heart chambers, which is especially important for cats, on the other hand, deep intertrabecular recesses in the ventricular cavity in combination with a reduced pumping function of the heart.
Diagnostics
The primary method for diagnosing LVNC is echocardiography. In the future, magnetic resonance and computed tomography can be used to clarify the diagnosis; contrast ventriculography is also used in humane medicine.
Echocardiographically, LV NML is represented by a two-layer structure of the ventricular myocardium - a thin epicardial compact layer and a trabecular, with deep recessions, non-compact layer, most often localized in the apical region and on the free wall of the left ventricle. There are several options for combining imaging criteria to make the diagnosis of LVNC. One of the most common is the ratio of the non-compact layer to the compact layer of more than 2 in adults and 1.4 in children, measured at the end of systole, the presence of several trabeculae in one section and deep recesses communicating with the cavity of the left ventricle, which is visualized by color Doppler mapping (Fig. .2).
Magnetic resonance imaging is a highly sensitive and specific diagnostic method for suspected LVNC, especially if the cardiac apex is not clearly visible during echocardiography. According to AHA recommendations, CT scanners with a power greater than 1.5 Tesla should be used to obtain the clearest images. The criterion for LVNC will be the presence of two layers of myocardium, and one of several diastolic sections along the long or short axes with the most pronounced trabecularity will be selected, while, unlike echocardiography, the ratio of the non-compact layer to the compact layer should be more than 2.3. A 17-segment heart model is used to determine localization.
Differential diagnosis
LVNC can be mistaken for various forms of HCM, since in both pathologies there may be hypertrophy of the free wall of the left ventricle and, most importantly, the apical region. The error in describing LVNC as DCM is due to the fact that systolic function may be moderately or significantly reduced, and the ventricular cavity is dilated. Errors in defining fibroelastosis and endomyocardial fibrosis are due to the fact that changes in these pathologies primarily affect the apical region, which is most difficult for visual assessment, and intertrabecular recesses can be poorly visualized during echocardiographic examination. In myocarditis, the ventricular walls may be thickened (in cats) and systolic function may be reduced (in dogs); similar changes can be found in LVNC.
According to Stöllberger and J. Finsterer, the most common misdiagnosis, in descending order, would be HCM, DCM, fibroelastosis, myocarditis, RCM and other causes.
Treatment and prognosis
Asymptomatic patients in the absence of rhythm and conduction disturbances do not require specific therapy. In other cases, it is no different from the usual pathogenetic and symptomatic treatment of chronic heart failure.
Predictors of an unfavorable prognosis will be dilatation of the heart chambers and severe rhythm and conduction disturbances.
Dynamic observation in all cases is recommended at least 2 times a year.
Clinical case
A 3-month-old Bengal kitten with radiographically detected cardiomegaly was admitted to an appointment with a general practitioner. The only complaint was difficult, “grunting” breathing. Appetite and exercise tolerance are completely preserved. Auscultation revealed a systolic murmur on the left, grade 2–3 out of 6.
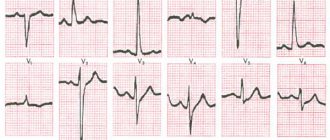
ECG – sinus rhythm, heart rate 208 per minute, no conduction disturbances were detected.
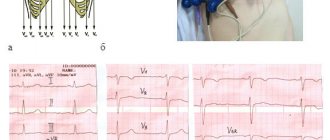
Echocardiography revealed increased trabecularity of the left ventricular apex with a characteristic “mosaic” blood flow (ratio of non-compact to compact layers in the range of 1.5–2.2) and apical mesoventricular obstruction with a gradient of 55 mm Hg. Art. (Fig. 3 and 4), moderate dilatation of the left atrium (13.7 mm, aorta 7 mm, short-axis measurements), grade 3–4 left ventricular diastolic dysfunction, ejection fraction 66% according to Simpson, severe pulmonary congestion circulation (PV/PVLA 1.5), moderate regurgitation on the mitral valve, no signs of pulmonary hypertension. The preliminary diagnosis based on the study data is unclassified cardiomyopathy, mesoventricular obstruction against the background of non-compact development of the left ventricle. Differential diagnosis: apical form of left ventricular hypertrophy. Of course, in this case, the diagnosis of LVNC is strictly preliminary and, given the extremely rare incidence of the disease, must be confirmed histologically.
Unfortunately, the owner of the animal did not show up for the follow-up appointment and nothing is known about the patient’s further fate. An interesting fact is that this kitten is the only survivor from the second litter. The first litter, five kittens in total, was entirely stillborn. The second litter also had five kittens: two of them were stillborn, two were killed by the cat immediately after birth, and the only surviving kitten 3 months later was admitted to a cardiologist with a rare myocardial disease.
Literature
1. Angelini P. Can Left Ventricular Noncompaction Be Acquired, and Can It Disappear? Texas Heart Institute Journal. 2017; 44 (4): 264–265. P.M.C. Web. 27 Oct. 2021.
2. Shemisa K. et al. Left Ventricular Noncompaction Cardiomyopathy. Cardiovascular Diagnosis and Therapy. 2013; 3 (3): 170–175. P.M.C. Web. 27 Oct. 2021.
3. Ali Sulafa KM Ventricular Noncompaction: Over or under Diagnosis? Journal of the Saudi Heart Association. 2009; 21 (3): 191–194. P.M.C. Web. 27 Oct. 2021.
4. Lin Ying-Nan et al. Left Ventricular Noncompaction Cardiomyopathy: A Case Report and Literature Review. International Journal of Clinical and Experimental Medicine. 2014; 7(12):5130–5133. Print.
5. Fazlinezhad A. et al. Echocardiographic Characteristics of Isolated Left Ventricular Noncompaction. ARYA Atherosclerosis. 2016; 12 (5): 243–247. Print.
6. Kittleson MD et al. Naturally Occurring Biventricular Noncompaction in an Adult Domestic Cat. Journal of Veterinary Internal Medicine. 2017; 31(2):527–531. P.M.C. Web. 27 Oct. 2017.
7. Zhang W. et al. Molecular Mechanism of Ventricular Trabeculation/compaction and the Pathogenesis of the Left Ventricular Noncompaction Cardiomyopathy (LVNC). American journal of medical genetics. Part C, Seminars in medical genetics. 2013; 163(3):144–156. P.M.C. Web. 27 Oct. 2021.
8. Weir-McCall, Jonathan R. et al. Left Ventricular Noncompaction: Anatomical Phenotype or Distinct Cardiomyopathy? Journal of the American College of Cardiology. 2016; 68(20):2157–2165. P.M.C. Web. 27 Oct. 2021.
9. Jenni R., Oechslin E., Schneider J. et al. Echocardiographic and pathoanatomical characteristics of isolated left ventricular non-compaction: a step towards classification as a distinct cardiomyopathy. 2001; 86:666–671.
10. Golukhova E.Z., Shomakhov R.A.
Non-compact myocardium of the left ventricle. Creative cardiology. 2013. Back to section
Treatment
The basis of therapeutic tactics in patients with spongy myocardium is the prevention and treatment of heart failure, heart rhythm disturbances and thromboembolic complications (taking anticoagulants for impaired left ventricular function). Asymptomatic patients and patients without arrhythmias and signs of left ventricular dysfunction are not prescribed treatment. Some patients are indicated for heart transplantation. Patients with malignant rhythm disorders are recommended to have a cardioverter-defibrillator implanted [1, 2, 7, 9].
The prognosis for patients with non-compact myocardium of the left ventricle depends on the volume of the affected segments, the overall contractility of the myocardium, the time of occurrence and the rate of increase of symptoms of heart failure. According to I. Jedlinsky and co-authors, mortality within 6 years was 50%. Of the 34 patients observed in the study by E. Oechslin et al., 12 patients died over a 44-month period, 6 of whom were diagnosed with sudden cardiac death, 4 with terminal heart failure, four patients underwent a heart transplant, and another four received a cardioverter-defibrillator. . The prognosis is especially poor in patients with an ejection fraction of less than 35%. Factors indicating an unfavorable prognosis of the disease include: large end-diastolic size of the left ventricle, persistent atrial fibrillation, and bundle branch block on the electrocardiogram.
During pathological studies , it was established that isolated non-compact myocardium is characterized by the presence of hypertrophied segments of the left ventricular wall, which consists of a non-compact endocardial layer, in which increased trabecularity is noted, and a thin epicardial compact layer. Deep intertrabecular recesses communicate with the cavity of the left ventricle, but not with the coronary blood flow, in contrast to non-compact myocardium associated with other congenital heart defects, when intertrabecular recesses communicate with the cavity of the left ventricle and the vessels of the coronary circulation (non-insulated myocardium) [1, 2, 8 , 10].
Symptoms
Although the disease can be diagnosed at any age, symptoms generally begin to appear in infancy. Some patients may have an undiagnosed form of LVHT, in such cases there are practically no symptoms.
The manifestations of LVH vary from one individual to another depending on the size and location of the non-consolidated myocardium. Symptoms are not strictly specific to LVHT, since they are often detected in other types of cardiomyopathy and diseases of the cardiovascular system, which often complicates diagnosis.
Clinical manifestations usually occur because:
- non-compact myocardium does not allow the heart to pump blood well throughout the body (which can cause symptoms of heart failure because the heart does not meet the body's needs);
- Myocardial trabecularity affects the normal electrical activity of the heart (which can cause arrhythmia, an abnormal heart rhythm).
Symptoms of LVH include:
- shortness of breath;
- fatigue (sometimes extremely pronounced);
- feeling dizzy;
- fainting;
- feeling of abnormal heartbeats (palpitations);
- swelling of the legs, ankles and feet (edema).
Clinical case of non-compact myocardium of the left ventricle in a 6-year-old girl
The child Melania B. was born from the first pregnancy, which occurred against the background of chronic maternal pyelonephritis and chronic intrauterine fetal hypoxia, from the first urgent birth at 39 weeks of gestation with a body weight of 3700 g. A short umbilical cord was noted during childbirth. On the 1st day after birth, a birth injury was diagnosed - rotational subluxation of the first cervical vertebra C1, which was clinically manifested by signs of movement disorder syndromes and vegetovisceral disorders. Neurosonography revealed ultrasound signs of grade I–II periventricular ischemia. Due to the presence of signs of post-hypoxic syndrome of maladjustment of the cardiovascular system, the girl underwent echocardiography, which revealed echo signs of a minor anomaly of heart development - an abnormal chord of the left ventricle, hypertrabecularity of the left and right ventricles, proliferation of papillary muscles, carditis.
From the maternity hospital, the girl was transferred to the neonatal pathology department. In addition to the listed examinations, echocardiography is provided on the 12th day of life: the heart chambers are not dilated, no pathological shunts are identified, chordal features of the left ventricle, systolic-diastolic function of the left ventricle is not changed. The electrocardiogram shows slowing of atrioventricular conduction, diffuse changes in the ventricular myocardium. After treatment, the girl was discharged home at the age of 1 month with recommendations for observation by a cardiologist, neurologist, and pediatrician.
Subsequently, the child’s condition gradually worsened, symptoms of heart failure increased, and therefore the girl at the age of 5 months was hospitalized in the cardio-rheumatology department of the Lugansk city multidisciplinary children's hospital No. 1 with a diagnosis of congenital non-rheumatic carditis of severe degree, decompensated, circulatory disorder of degree IIA, thymomegaly II degree, minor cardiac anomaly, motor impairment syndrome, perinatally caused. When examined on an electrocardiogram, there is dilatation of the left ventricle with systolic overload, diffuse changes in the myocardium. A repeat electrocardiogram 4 days later shows dilatation of the left ventricle and left atrium with secondary mitral insufficiency of degree IIB, possibly due to previous myocarditis, and a decrease in the systolic and diastolic function of the left ventricle. An echocardiogram after 9 days showed moderate dilation of the left chambers of the heart, decreased contractility of the left ventricular myocardium, and moderate individual increased trabecularity of the left ventricle. Consultation with a cardiac surgeon: there is no heart defect. In the department, the child was prescribed digoxin for the first time. The girl was discharged from the department at the insistence of her parents at the age of 5 months 13 days.
At the age of 8 months, the girl’s condition worsened; she was again hospitalized in the cardio-rheumatology department with a diagnosis of congenital non-rheumatic carditis, severe. Stage of decompensation, circulatory disorder IIB degree, relative insufficiency of the mitral and tricuspid valves, slowdown of statokinetic function, rickets II degree, subacute course, malnutrition I degree, immunodeficiency state. Two weeks later the girl was discharged home with improved condition.
Subsequently, the child was repeatedly consulted by a cardiologist, examined, and received treatment. An electrocardiogram at the age of 9 months revealed signs of incomplete blockade of the right bundle branch.
At the age of 1 year 3 months, the girl was examined and consulted in the scientific and practical medical center of pediatric cardiology and cardiac surgery in Kiev, where the diagnosis was made: cardiomyopathy, non-compact myocardium of the left ventricle, severe mitral regurgitation, high pulmonary hypertension. Recommendations were given for further tactics of examination and treatment of the patient. Subsequently, the girl was regularly examined and treated at the specified center.
Cardiac magnetic resonance imaging without intravenous contrast was performed at 3 years and 2 months of age. A series of tomograms show enlargement of the cavities of both ventricles (the size of the left ventricle is 5.6 x 3.6 x 3.6 cm, the right ventricle is 4.4 x 2.25 cm), the left atrium is significantly enlarged, and moderate insufficiency of the atrioventricular valves is observed. The endocardium of the left ventricle has an open trabecular structure with a relative thinning of the compact subepicardial part up to 0.25 cm. The diastolic ratio of the non-compact and compact parts of the left ventricle is 1.25/0.25 cm. Similar structural changes in the myocardium are determined in the right ventricle. Left ventricular ejection fraction - 27%. A small amount of fluid in the pericardial cavity. The girl has not been genetically examined.
Due to deterioration of her condition, the girl was again referred for treatment at the age of 5 years and 3 months. Diagnosis: non-compact myocardium of the left ventricle. Severe tricuspid and mitral valve insufficiency, dilatation of the left heart, severe pulmonary hypertension. An echocardiogram shows the left chambers of the heart are dilated, myocardial contractility is satisfactory. Pronounced trabecularity of the posterior wall of the left ventricle, the thickness of the compact part is 2.5 mm, the non-compact part is 12 mm. Conclusion: non-compact myocardium of the left ventricle, insufficiency of the tricuspid and mitral valves, dilatation of the left chambers of the heart, severe pulmonary hypertension. Repeated echocardiography after 10 days of treatment revealed: the left chambers of the heart are enlarged, myocardial contractility is good (positive dynamics during treatment). Diastolic disorders of the left ventricle type III (restrictive). Severe trabecularity of the left ventricle (non-compact myocardium). There is no fluid in the pericardial cavity and pleural cavities. During the treatment period, the girl received dobutamine, Lasix, asparkam, digoxin, berlipril, panangin, and veroshpiron. Considering the positive dynamics of the patient’s clinical condition, the council of doctors decided that surgical treatment was not indicated and it was recommended to continue treatment on an outpatient basis. Currently, the girl's condition is compensated.